
一测多评法同时测定新复方大青叶片中4 种成分含量*
2023-05-26 10:54王丽琼魏长勇余敏灵
中国药业 2023年10期
王丽琼,魏长勇,余敏灵
(四川省乐山市食品药品检验检测中心·四川省药品监督管理局中药质量研究重点实验室,四川 乐山 614000)
新复方大青叶片是由复方大青叶提取物、对乙酰氨基酚、咖啡因、异戊巴比妥和维生素C 组成的复方制剂。现行质量标准为原国家食品药品监督管理总局国家药品标准(WB3-B-1057-91-2015)[1],但因维生素C 对照品性质不稳定[2-5],未对其含量进行控制,且咖啡因和异戊巴比妥对照品存在需特殊购买和管理的问题。为此,本研究中采用乙醇代替甲醇(乙腈)作为流动相的有机相,建立了以对乙酰氨基酚为对照品,同时测定新复方大青叶片中4 种成分的一测多评(QAMS)法。现报道如下。
1 仪器与试药
1.1 仪器
1260 型高效液相色谱仪[含二极管阵列(DAD)检测器]、1100 型高效液相色谱仪(含紫外- 可见光检测器),均购自美国Agilent 公司;PE A-10 型高效液相色谱仪,含量DAD 检测器(珀金埃尔默企业管理有限公司);XS205 型电子分析天平(瑞士Mettler Toledo 公司,精度为0.01 mg);KQ 5200DB 型数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药
新复方大青叶片(厂家S1,S2,S3,S4,S5,编号分别为A,B;C,D;E;F;G,规格均为每片含维生素C 10 mg、对乙酰氨基酚75 mg、咖啡因7.5 mg 和异戊巴比妥7.5 mg);维生素C 对照品、对乙酰氨基酚对照品、咖啡因对照品(中国食品药品检定研究院,批号分别为100425 - 201504,100018 - 201610,171215 - 201010,含量分别为100.0%,99.9%,100.0%);异戊巴比妥原料(厂家A,批号为W201102,含量为99.4%);乙醇为色谱纯,磷酸、乙醇、磷酸二氢钾、焦亚硫酸钠均为分析纯,水为纯化水。
2 方法与结果
2.1 色谱条件
色谱柱:Welch Ultimate LP - C18柱(250 mm ×4.6 mm,5 μm);流动相:乙醇(A)-0.01 mol/L 磷酸二氢钾溶液(B,用磷酸调pH 至3.0),梯度洗脱(0~3 min时2%A,3~6 min 时2%A→15%A,6~15 min 时15%A,15~20 min 时15%A→60%A,20~26 min 时60%A,26~27 min 时60%A→2%A,27~32 min 时2%A);流速:0.8 mL/min;检测波长:215 nm(对乙酰氨基酚、咖啡因和异戊巴比妥)和245 nm(维生素C);柱温:40 ℃;进样量:10 μL。
2.2 溶液制备
分别取维生素C、对乙酰氨基酚、咖啡因对照品和异戊巴比妥原料32.16,187.06,18.66,19.06 mg,精密称定,置同一50 mL容量瓶中,加少量无水乙醇溶解,再加0.5%焦亚硫酸钠溶液(用磷酸调pH 至3.0,下同)稀释、定容、摇匀,即得混合对照品贮备液;精密量取1 mL,置20 mL 容量瓶中,加0.5%焦亚硫酸钠溶液稀释、定容,摇匀,即得混合对照品溶液。取样品20片(糖衣片除去包衣),研细,取约0.1 g,精密称定,置100 mL 容量瓶中,加0.5%焦亚硫酸钠溶液适量,超声(功率200 W、频率40 kHz,下同)处理10 min,再加0.5%焦亚硫酸钠溶液稀释、定容、摇匀,即得供试品溶液。
2.3 方法学考察
系统适用性试验:取2.2项下混合对照品溶液和供试品溶液各适量,按2.1 项下色谱条件进样测定,记录色谱图。结果供试品溶液色谱中,在与混合对照品溶液色谱相应位置处有吸收峰,理论板数按各成分峰计均应大于3 000,分离度均大于1.5。详见图1。

图1 高效液相色谱图1.Vitamin C 2.Paracetamol 3.Caffeine 4.AmobarbitalA.Mixed reference solution B.Test solutionFig.1 HPLC chromatograms
线性关系考察:精密量取2.2项下混合对照品溶液2,4,8,12,16,20 μL,按2.1项下色谱条件进样测定,记录峰面积。以待测成分进样量(C,μg)为横坐标、峰面积(A)为纵坐标进行线性回归,得维生素C、对乙酰氨基酚、咖啡因、异戊巴比妥的回归方程分别为A1=3 627.24C1(r=0.999 8),A2=2 036.51C2(r=0.999 7),A3=5 570.84C3(r=0.999 9),A4=2 467.71C4(r=0.999 9)。结果表明,4种成分进样量分别在0.064 3~0.643 2 μg、0.373 7~3.737 5 μg、0.037 3~0.373 2 μg、0.037 9~0.378 9 μg范围内与峰面积线性关系良好。
检测限与定量限考察:取维生素C、对乙酰氨基酚、咖啡因和异戊巴比妥对照品各适量,精密称定,加无水乙醇,制成质量浓度分别为1.21,7.48,3.81,8.17 μg/mL的溶液,按2.1项下色谱条件进样测定,记录峰面积,以信噪比(S/N)为3和10时的待测成分质量浓度分别记作检测限和定量限。结果检测限分别为0.03,0.15,0.07,0.24 μg/ mL,定量限分别为0.09,0.51,0.23,0.79 μg/mL。
精密度试验:取2.2 项下混合对照品溶液适量,按2.1项下色谱条件连续进样测定6次,记录峰面积。结果维生素C、对乙酰氨基酚、咖啡因和异戊巴比妥峰面积的RSD分别为1.02%,0.92%,1.09%,0.78%(n= 6),表明仪器精密度良好。
稳定性试验:取2.2 项下供试品溶液适量,分别于室温下放置0,2,4,8,16,24 h时按2.1项下色谱条件进样测定,记录峰面积。结果维生素C、对乙酰氨基酚、咖啡因和异戊巴比妥峰面积的RSD分别为1.58%,1.08%,0.97%,0.85%(n=6),表明供试品溶液在室温下放置24 h内基本稳定。
重复性试验:取样品(编号A)适量,按2.2项下方法制备供试品溶液,共6 份,按2.1 项下色谱条件进样测定,记录峰面积并计算含量。结果维生素C、对乙酰氨基酚、咖啡因和异戊巴比妥的平均含量分别为70.31%,89.37%,93.87%,89.72%,RSD分别为1.50%,1.72%,0.79%,1.21%(n=6),表明方法重复性良好。
加样回收试验:取已知含量的样品(编号A,维生素C、对乙酰氨基酚、咖啡因和异戊巴比妥的平均含量分别为22.461 0,217.828 8,21.985 1,21.840 2 mg/ g),取细粉各适量(约0.05 g),共6份,置100 mL容量瓶中,精密加入2.2 项下混合对照品贮备液2.5 mL,按2.2 项下方法制备供试品溶液。按2.1 项下色谱条件进样测定,记录峰面积,并采用外标法(ESM)计算回收率。结果见表1。

表1 加样回收试验结果(n=6)Tab.1 Results of the recovery test(n=6)
2.4 相对校正因子(f)的确定和耐用性考察
f的确定:精密量取2.2 项下混合对照品溶液适量,共3 份,按2.1 项下色谱条件进样测定,记录峰面积。以对乙酰氨基酚为参照,采用斜率法计算维生素C、咖啡因和异戊巴比妥的f,f=Kr/Kt,式中,Kr为对乙酰氨基酚的斜率,Kt为其他待测成分的斜率。计算3 个成分f的平均值。结果维生素C、咖啡因和异戊巴比妥的f分别为0.576 1,0.373 2,0.843 8,RSD分别为0.91%,0.51%,0.62%(n=3)。
耐用性考察:精密量取2.2项下混合对照品溶液适量,按2.1项下色谱条件[分别设置色谱柱为Agilent Extend - C18柱、Welch Ultimate LP - C18柱、A585V3 SVEATMC18Opal 柱、GL Sciences Intersustain AQ - C18柱、Waters AtlantisTMT3 柱(规格均为250 mm × 4.6 mm,5 μm)]测定,记录峰面积,计算维生素C、咖啡因和异戊巴比妥的f,结果见表2。可见,色谱柱发生一定程度变化时,各成分f的RSD均小于3%,耐用性较好。
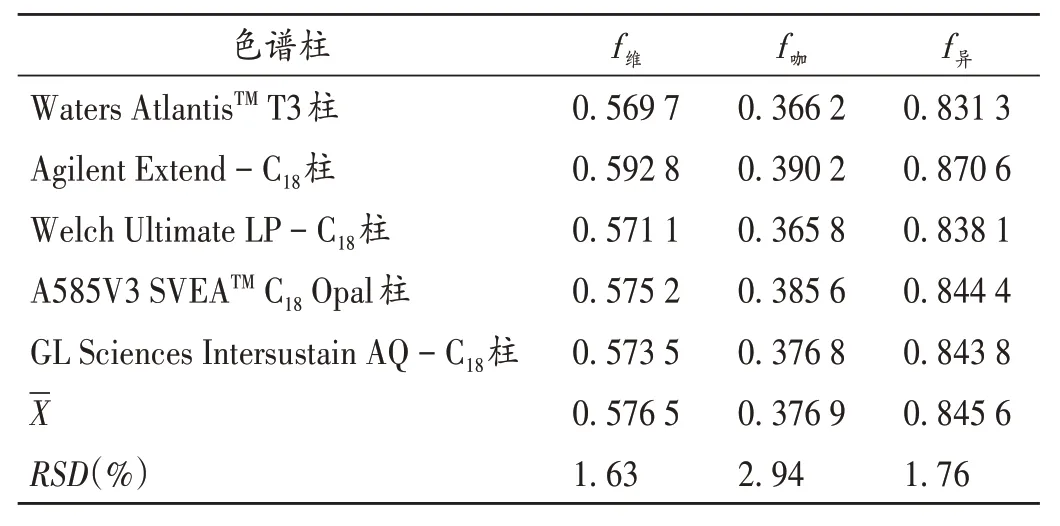
表2 耐用性考察结果Tab.2 Results of the durability test
2.5 QAMS 法与外标法测定结果比较
取样品(编号A - G)适量,按2.2 项下方法制备供试品溶液,均平行3 份,按2.1 项下色谱条件进样测定,记录峰面积,分别采用ESM 和QAMS 法计算样品含量。QAMS法计算公式为mt=(f×At×As)/ms,式中,mt为待测成分(维生素C、咖啡因和异戊巴比妥)的质量(μg),At为式中待测成分的峰面积,ms与As分别为对乙酰氨基酚的质量(μg)与峰面积。对2种方法所得结果采用配对t检验,结果均无显著差异(P>0.05),详见表3。

表3 样品含量测定结果(%)Tab.3 Results of content determination of vitamin C,caffeine,amobarbital and paracetamol in samples(%)
3 讨论
3.1 样品溶剂选择
预试验中比较了0.5%焦亚硫酸钠溶液及0.5%焦亚硫酸钠溶液-乙醇(95∶5、90∶10、80∶20,V/V)这4 种溶剂对4个成分提取效率的影响。结果,4种溶剂提取效率基本一致,乙醇对对乙酰氨基酚、咖啡因、异戊巴比妥的提取均无影响,综合考虑,溶剂最终选择0.5%焦亚硫酸钠溶液。
3.2 检测波长选择
预试验中所得紫外光谱图显示,维生素C在245 nm波长处有最大吸收,咖啡因和异戊巴比妥在215 nm 波长处有较大吸收,综合考虑,选择检测波长为215 nm(对乙酰氨基酚、咖啡因和异戊巴比妥)和245 nm(维生素C)。
3.3 流动相与柱温选择
预试验中比较了流动相B分别为水、0.01%磷酸溶液、0.01 mol/L 磷酸二氢钾溶液(用磷酸调pH 至3.0)对色谱峰的影响。结果采用水作为流动相时,维生素C出峰时间延迟,峰形差,展宽,而在酸性条件下,维生素C出峰时间适中,峰形好。经考察,选择0.01 mol/L 磷酸二氢钾溶液(用磷酸调pH 至3.0)时,4个成分峰与相邻峰的分离度符合要求。4 种成分理化性质不一致,维生素C极性强,异戊巴比妥非极性强,采用梯度洗脱,各组分出峰时间适中,峰间分离度好。乙醇黏度较大,适当升高柱温可降低黏度,经考察,柱温选择40 ℃。
3.4 维生素C 稳定性考察
预试验中以维生素C 对照品加磷酸水溶液(pH 分别为2.0,2.5,3.0,3.5,4.0,4.5)及水,分别于室温下放置0,2,8,12,16,24,36 h 时进样测定,记录峰面积。发现维生素C 溶液稳定性pH 3.0 >pH 3.5 >pH 2.5 >pH 4.0 >pH 4.5 >pH 2.0 >水。故选择制备pH 3.0 的磷酸水溶液。还分别用0.3%,0.5%,0.7%焦亚硫酸钠溶液制备供试品溶液,分别于0,4,8,12,16,24 h 时进样测定。结果发现焦亚硫酸钠溶液体积分数为0.5%及以上时,维生素C稳定性较好。
3.5 方法的经济性与环保性
新复方大青叶片各成分中,异戊巴比妥和咖啡因属第二类精神药品,维生素C 性质不稳定(易氧化和水解),这3个成分对照品的购买和管理存在一定的问题。QAMS 法是以价廉易得、性质稳定的物质为内参物,建立内参物与其他成分间的f,并以此获得其他多个组分的含量[6-9]。该方法可解决药品质量控制中对照品性质不稳定、价格昂贵、需特殊购买和管理的问题,适用于复杂成分或多指标成分含量测定。课题组经过研究和验证,以性质稳定的对乙酰氨基酚为内参物,采用QAMS 法[10-16],以f计算样品中维生素C、咖啡因和异戊巴比妥的含量。在后期检验过程中,仅需对乙酰氨基酚对照品,省去维生素C、咖啡因和异戊巴比妥对照品,实现检验方法的经济性发展。甲醇作为第二类溶剂在检验中被大量使用,对环境和实验人员身体健康影响均较大。乙醇属第三类溶剂,毒性较低,本研究中以乙醇作为流动相的组成部分较安全[17-20]。
3.6 方法评价
本研究中所建立的方法环保(以乙醇代替甲醇)、经济(只需1 种对照品),结果准确且可快速获得,适用于新复方大青叶片的质量评价。
猜你喜欢
山西化工(2021年5期)2021-01-25
硫酸工业(2020年10期)2020-12-10
保健与生活(2020年4期)2020-03-02
中国外汇(2019年10期)2019-08-27
中国外汇(2019年10期)2019-08-27
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
天然产物研究与开发(2018年5期)2018-06-13
癌变·畸变·突变(2016年3期)2016-02-27
中国药业(2014年24期)2014-05-26
