
髓系细胞触发受体-1在冠状动脉粥样硬化性心脏病中的研究进展
2023-05-12 06:30蒋梦婷张瑜高磊
心血管病学进展 2023年4期
蒋梦婷 张瑜 高磊
(1.中国人民解放军总医院第六医学中心心血管病医学部,北京 100853; 2.天津中医药大学临床实训教学部,天津 301617)
髓系细胞触发受体(triggering receptor expressed on myeloid cells,TREM)是一种免疫球蛋白超家族活化受体,主要表达于多种髓系细胞,在非髓系的上皮细胞和内皮细胞上也有表达[1]。TREM-1是首个被发现的TREM家族成员,能促进微生物产物对免疫细胞的激活,被认为是“炎症放大器”。随着研究的深入,发现TREM-1在冠心病发生和发展的各个阶段发挥着重要作用。现对TREM-1的研究现状及其在冠心病中的研究进展做一综述,探讨其作为冠心病治疗靶标的可能性。
1 TREM-1概述
1.1 TREM-1的分布及结构
TREM-1是一种相对分子质量3.0×104的糖蛋白表面受体,位于染色体6p21.1上,广泛分布于成熟的单核细胞、巨噬细胞、中性粒细胞等髓系细胞表面,也可见于上皮细胞、内皮细胞等非髓系细胞表面[2-3]。与其余跨膜糖蛋白类似,TREM-1包含三个部分:细胞外免疫球蛋白结构域、跨膜区和短胞质结构域。其胞外结构域具有免疫球蛋白型折叠,因此属于免疫球蛋白超家族。其胞质结构域中仅包含5个氨基酸,没有信号序列,跨膜区与含有免疫受体酪氨酸激活模体(immunoreceptor tyrosine-based activation motif,ITAM)的DNAX相关蛋白12(DNAX-associated protein 12,DAP12)(一种跨膜适配器,在多种细胞中发挥传递炎症信号的作用)结合,以表达其功能。虽然TREM-1分子的基本形式是单体,但它在细胞膜上的活化和稳定依赖于多聚化[4]。
1.2 TREM-1的可溶形式
同多种膜受体一样,TREM-1也有其可溶形式。可溶性髓系细胞触发受体-1(soluble triggering receptor expressed on myeloid cells-1,sTREM-1)分子结构和TREM-1胞外区相似,但并不具备跨膜区和短胞质结构域,主要由基质金属蛋白酶(matrix metalloproteinase,MMP),如MMP-9,对锚定的TREM-1蛋白水解切割产生。可与TREM-1竞争性结合配体,具有一定的抑制TREM-1信号转导的作用[5]。sTREM-1是TREM-1受体激活的标志,与TREM-1通路活性相关[6]。目前血清sTREM-1在炎症相关疾病中的释放已被广泛研究,一些临床研究[7-8]认为sTREM-1可作为感染性疾病预后的可靠生物标志物,最近的研究[9]发现了其与新型冠状病毒肺炎患者预后的相关性。
1.3 TREM-1抑制剂
TREM-1可以在蛋白质和转录水平被抑制。蛋白质水平的TREM-1抑制剂主要指干扰TREM-1配体相互作用的小肽,如LR-12、LP-17。这些抑制剂可以以游离形式递送[1,10],也可以通过慢病毒载体系统[11]。转录水平的TREM-1抑制剂主要包括小干扰RNA、短发夹RNA等,通过干扰TREM-1基因抑制TREM-1表达[12-13]。
2 TREM-1的作用机制
2.1 TREM-1与DAP12信号通路
TREM-1的激活在不同细胞的机制有所差异。在单核细胞表面,配体与TREM-1结合,TREM-1上调表达并在细胞表面聚集发生多聚化,继而MMP水解切割部分sTREM-1释放;在中性粒细胞表面则表现为细胞膜上TREM-1的快速重组。激活的TREM-1的跨膜区含有一个赖氨酸残基217,与DAP12中的天冬氨酸相互作用并转导下游信号[4]。随后,DAP12的ITAM磷酸化,激活非受体型脾酪氨酸激酶(spleen tyrosine kinase,SYK),激活磷脂酰肌醇3激酶(phosphatidylinositol 3-kinase,PI3K)、磷脂酶C(phospholipase C,PLC)、胞外信号调节激酶(extracellular signal-regulated kinases,ERK)1/2、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)、核转录因子-κB(nuclear factor-κB,NF-κB),增加炎症基因的表达以及活性氧的释放,促进了白细胞介素(interleukin,IL)-1β、IL-6、IL-8、肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、单核细胞趋化蛋白-1(monocyte chemoattractant protein-1,MCP-1)和粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony-stimulating factor,GM-CSF)等炎症因子的释放[14-16]。另外,也有研究[16]发现SYK磷酸化能够调节钙离子内流从而激活MAPK/ERK途径。
2.2 TREM-1与Toll样受体及NOD样受体的协同作用
单独激活TREM-1不会诱导持续的炎症反应,表明TREM-1依赖于与其他途径的协同作用。目前已发现TREM-1与胞外模式识别的Toll样受体(Toll-like receptor,TLR)和胞内模式识别的NOD样受体(nucleotide-binding oligomerization domain-like receptor,NLR)具有协同效应,在免疫反应中促进了细胞因子诱导的炎症。
TLR及NLR均是模式识别受体,在病原相关分子模式或损伤相关分子模式帮助下刺激炎症反应,是参与非特异性免疫的一类重要分子[17-18]。目前已发现了部分TREM-1与TLR的协同作用,如TREM-1与TLR-4相互作用,募集炎症介质,如PI3K、ERK 1/2、白细胞介素-1受体相关激酶1(interleukin-1 receptor associated kinase-1,IRAK-1)和NF-κB,诱导暴露于脂多糖的离体细胞产生更多炎性细胞因子[16]。另有一些研究[19]发现了TREM-1与NLR的协同作用,可能是通过激活PI3K和/或p38/MAPK通路,使TNF-α、IL-1β和IL-6的表达上调。
2.3 TREM-1与细胞自噬-凋亡
细胞自噬是细胞在缺氧、炎症等状态下,通过对抗破坏细胞稳态的内质网应激,减少炎性反应,使细胞重返稳态的过程。最新研究[13]发现,TREM-1过表达可引起自噬基因Beclin-1、Atg-5、LC3b及抗凋亡基因表达下调,促凋亡基因表达上调。用TREM-1抑制剂处理结肠炎小鼠,能够抑制内质网应激引发的未折叠蛋白反应和自噬水平的提高[20]。这些研究均提示TREM-1可能对细胞自噬起负性调节作用。
2.4 TREM-1与巨噬细胞极化
TREM-1在多系统模型中均表现出促进巨噬细胞M1型极化的作用。在消化系统中,TREM-1缺乏导致分泌IL-1β的M1巨噬细胞数量减少[21]。在糖尿病肾病大鼠中,TREM-1与M1巨噬细胞极化标志物诱导型一氧化氮合酶呈正向变化,且维生素D可降低高血糖大鼠的TREM-1及诱导型一氧化氮合酶水平[22]。Wang等[23]用脂多糖处理的小鼠气道上皮细胞分离出的外泌体诱导了M1巨噬细胞极化,且极化可被TREM-1敲除部分逆转,提示了TREM-1诱导的巨噬细胞极化与外泌体的相关性。
2.5 TREM-1与细胞焦亡
细胞焦亡是指细胞炎性形式的程序性死亡。近期两项研究报道了TREM-1与细胞焦亡的关系。NOD样受体蛋白3(NOD-like receptor protein 3,NLRP3)是NLR家族的成员,NLRP3炎症小体可被SYK依赖性活性氧激活,是细胞焦亡的关键激活因子[24]。TREM-1抑制剂LP17降低了NLRP3炎症小体生成和炎症因子IL-1β、IL-18的水平,减少了焦亡相关蛋白caspase-1、GSDMD和GSDMD-N的表达,提示TREM-1可通过激活NLRP3炎症小体介导的焦亡,促进炎症反应[25]。TREM-1受体在斑块及细胞中的作用示意图如图1。
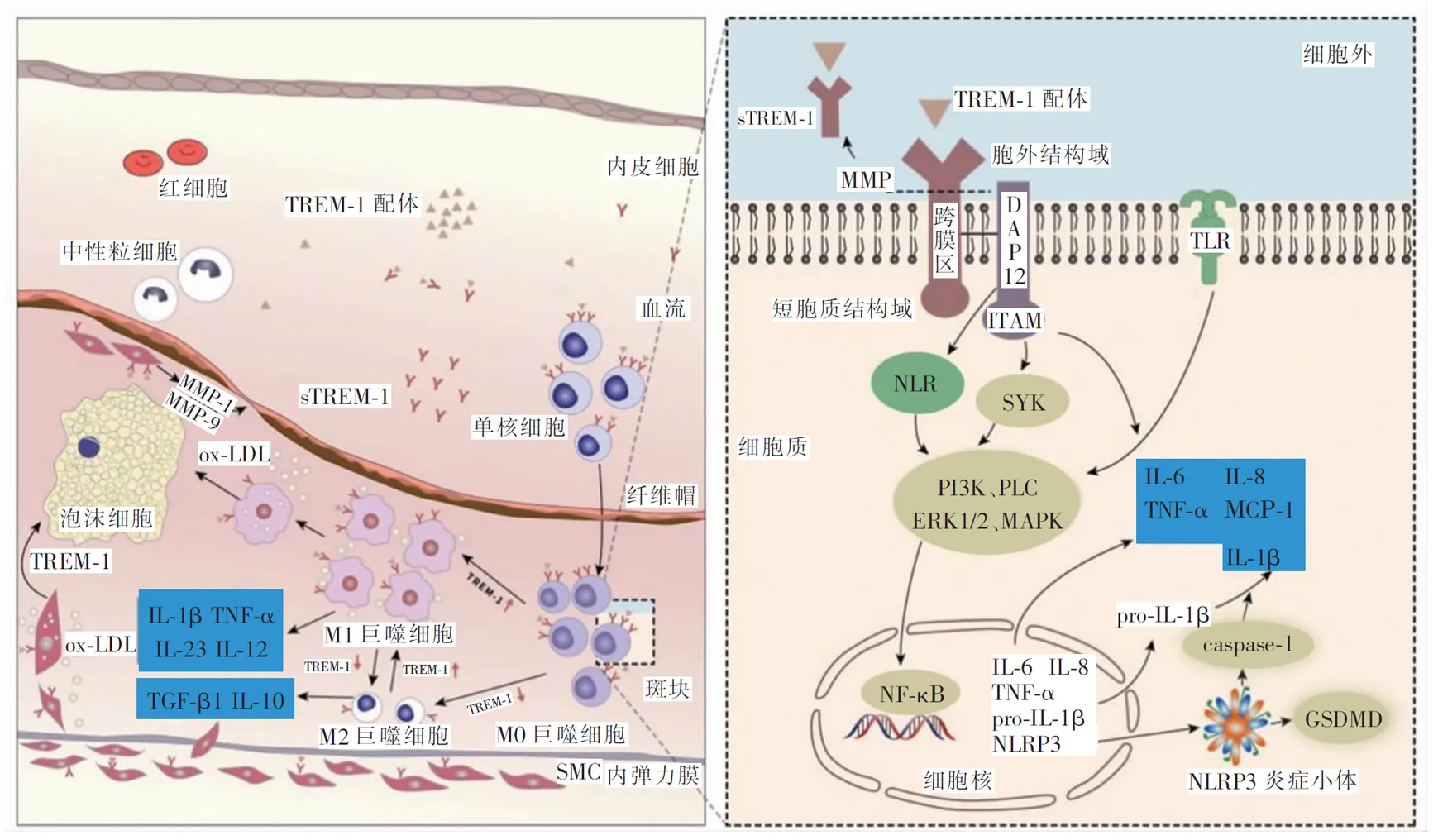
注:TGF-β1,转化生长因子-β1;ox-LDL,氧化低密度脂蛋白;SMC,平滑肌细胞;caspase-1,含半胱氨酸的天冬氨酸蛋白水解酶;GSDMD,消皮素D。
3 TREM-1在冠心病发生发展中的作用
3.1 TREM-1与动脉粥样硬化形成
动脉粥样硬化(atherosclerosis,AS)是一种慢性炎症性疾病,以动脉壁内脂质沉积为特征,氧化低密度脂蛋白(oxidized low-density lipoprotein,ox-LDL)是AS形成的重要促进因素。TREM-1在AS中的作用最早由Joffre等[1]阐述,他们发现下调TREM-1后,斑块的体积明显下降,TREM-1可促进巨噬细胞分泌大量炎症因子,转化为泡沫细胞,进而促进AS发展。进一步研究发现了ox-LDL在其中的关键作用。ox-LDL可上调TREM-1的表达,并且增强了TLR通路的炎症反应,导致细胞焦亡增加和自噬减少,从而促进AS发展[26-27],降脂药物普伐他汀可通过降低ox-LDL使AS斑块中TREM-1、DAP12和下游炎症因子TNF-α、IL-1表达降低,减少泡沫细胞形成[28]。sTREM-1血清浓度较高的个体体重指数、甘油三酯、低密度脂蛋白胆固醇值较高[29],也提示了TREM-1的激活与血脂的密切关系。此外,TREM-1基因多态性与AS严重程度相关,Kutikhin等[30]研究发现,TREM-1基因rs4711668的T/T基因型与重度AS显著相关。因此,近年来,TREM-1阻断剂LR-12和LP-17作为人工合成的小分子肽,成为冠心病治疗研究的热点,其主要机制可能是通过阻断TREM-1的多聚化和激活,干扰其与配体的结合。
3.2 TREM-1与斑块稳定性
典型的AS易损斑块表现为薄纤维帽粥样硬化斑块,主要由大的坏死核心和薄层纤维帽组成。TREM-1促进巨噬细胞向泡沫细胞分化,增大脂质核心[1]。斑块中的TREM-1诱导巨噬细胞和平滑肌细胞(smooth muscle cells,SMC)分泌MMP-1和MMP-9,它们可水解胶原,使纤维帽变薄[31]。此外,与无症状斑块相比,有症状斑块中TREM-1和DAP12上调,且有症状斑块来源的血管SMC比无症状斑块来源的SMC表达更高的TREM-1水平[32]。这些均表明,TREM-1可能导致斑块不稳定,加速斑块破裂。
3.3 TREM-1与急性冠脉综合征
当斑块发生破裂,覆盖坏死核心的纤维帽产生裂隙,脂质及坏死碎片暴露于血液中,继发血栓形成,阻塞血管管腔,引起急性冠脉综合征(acute coronary syndrome,ACS)。管腔阻塞导致的缺血缺氧导致大量模式识别受体释放,一些如HMGB1、HSP70、CD177、PGLYTRP1等作为配体与TREM-1结合,引起和放大炎症反应,产生的致炎细胞因子和趋化因子,又进一步引起中性粒细胞和单核细胞在梗死区聚集。此外,越来越多的证据提示,血浆sTREM-1水平是影响ACS预后的关键介质。血浆sTREM-1水平显著升高,可能是区分ACS和非特异性胸痛的生物标志物[33],其升高与全因死亡率和主要不良心血管事件风险增加相关[34]。在心源性休克患者中,非幸存者的sTREM-1血浆浓度高于幸存者[35]。值得注意的是,FAST-MI研究[36]的最新结果表明,尽管sTREM-1与死亡、心肌梗死复发和卒中风险增加有关,但未发现其血浆遗传水平是ACS患者预后的影响因素。
3.4 TREM-1与支架内再狭窄
支架内再狭窄(in-tent restenosis,ISR)指支架植入后支架内或者支架两端发生的超过50%的狭窄,其发生率为10%[37],是AS治疗的难题之一。在1 683例接受冠状动脉介入治疗和大约一年后随访并造影的患者中,130例被诊断为ISR,150例性别和年龄匹配的无ISR患者被随机纳入对照,发现与对照组相比,ISR组患者中sTREM-1的表达明显更高。体外试验也表明,TREM-1激活可促进血管SMC的炎症、增殖和迁移,从而促进其向支架部位募集和向泡沫细胞转化,引起新生动脉粥样硬化[2,38],正是ISR的成因之一。
3.5 TREM-1与缺血再灌注损伤
溶栓和冠状动脉介入技术极大地提高了ACS患者的生存率,但短时间内发生的缺血再灌注会加重心肌损伤,引起梗死面积增大、心律不齐、心脏功能障碍等。Lemarié等[39]在冠状动脉闭塞-再灌注技术诱导的猪心肌梗死模型中,通过抑制TREM-1,显著改善了平均动脉压、心脏指数、射血分数等心功能评估指标,改善了心脏功能,提示了TREM-1抑制剂在缺血再灌注改善方面的可能性。
总的来说,TREM-1通过其在AS分子通路中的关键位置,充当炎症、脂质积聚和斑块不稳定的放大器,是潜在的AS治疗靶点。TREM-1抑制作为抗AS治疗的主要理论优势是它提供了足够的抗炎作用,而没有显著干扰重要的免疫反应[40]。需要进一步的研究证明TREM-1在AS早期和晚期的意义,以及更多临床研究证明TREM-1抑制剂的抗AS作用。
4 总结与展望
TREM-1作为机体的“炎症放大器”,近年来引起了医疗行业的广泛关注。TREM-1的特异性抑制剂LR-12已作为治疗脓毒性休克的药物获美国食品和药物管理局快速通道资格。TREM-1在冠心病各个阶段的作用正在逐步揭开,更多的分子、动物和临床研究正在进行,围绕着TREM-1的多种药物也在开发。随着研究的进一步深入,TREM-1相关药物有望成为冠心病治疗的新星。目前,TREM-1抑制剂尚未在临床用于冠心病治疗,且暂无相关研究正在进行,对于药物的给药方式、药代动力学以及是否会对其他系统产生影响,尚需进一步论证。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
医学综述(2022年7期)2022-04-19
自我保健(2021年2期)2021-11-30
妇女之友(2021年9期)2021-09-26
昆明医科大学学报(2020年12期)2021-01-26
昆明医科大学学报(2020年11期)2020-12-28
世界科学技术-中医药现代化(2020年2期)2020-07-25
百姓生活(2019年2期)2019-03-20
上海农业学报(2017年3期)2017-04-10
中国当代医药(2015年16期)2015-03-01
