
基于深度神经网络对抗乳腺癌候选药物ERα生物活性的预测
2023-04-29 00:00:00花蕊,朱家明
陕西理工大学学报(自然科学版) 2023年2期




摘要: 针对抗乳腺癌候选药物的ERα生物活性建立定量结构-活性关系模型预测药物化合物的生物活性,首先通过主成分分析法,以化合物的生物活性值(IC50)为因变量,729个分子描述符为自变量。再利用皮尔逊相关系数,剔除高度相关的变量,最终得到20个最具有显著影响的变量。使用支持向量机(SVM)和深度神经网络(DNN)两种模型分别建立化合物对ERα生物活性定量预测模型,利用评价指标对两模型结果进行比较。结果显示DNN模型的预测结果较好,其均方根误差为0.735 5,均方误差为0.535 4,平均绝对百分比误差为0.086 1。研究得出的预测模型可以极大地节省药物研发时间,为新型抗乳腺癌先导化合物的药物研究提供实验和理论支持。
关键词: 乳腺癌药物;ERα生物活性;主成分分析法;支持向量机;深度神经网络;预测
中图分类号: TP391.4;R965.1" 文献标识码: A" 文章编号: 2096-3998(2023)02-0047-07
众所周知,乳腺癌是目前研究较多的恶性肿瘤之一,在女性癌症中的发病率位居首位,然而乳腺癌的早发现、早诊断、早治疗以及多种治疗手段相结合的多学科综合治疗的发展,使其死亡率呈下降趋势[1]。雌激素受体α亚型(ERα)与乳腺癌疾病的发生有着明显的关系,比如,临床治疗乳腺癌的经典药物他莫昔芬和雷诺昔芬就是ERα拮抗剂[2]。抗激素治疗常用于ERα表达的乳腺癌患者,其主要通过调节雌激素受体活性来控制体内雌激素水平,也就是能够拮抗ERα活性的化合物可能是治疗乳腺癌的候选药物[3]。因此,需要建立定量结构-活性关系模型来剔除大量的与化合物相关性弱的分子,去除这些分子后建立定量预测模型,得到预测结果更好的模型,再通过新化合物的IC50值分析新化合物的生物活性。
近年来,关于药物与靶点相互作用关系预测的模型报道越来越多,这些模型大多数是判断药物与靶点之间是否存在相互作用关系,只有少数模型用于预测药物与靶点之间的定量关系。这些定量模型预测性能较差且只是针对少量靶点,即模型的准确性和适用范围还需进一步提高。因此建立预测性能高与适用范围广的药物与靶点相互作用关系的定量预测模型是研究药物化学成分潜在作用靶点及活性预测有待解决的问题。
1数据来源与假设
本文数据来源于2021年“华为杯”数学建模竞赛D题,参考附件对所给数据进行预处理。根据文件“Molecular_Descriptor.xlsx”和“ERα_activity.xlsx”提供的数据,针对1974个化合物的729个分子描述符数据进行变量选择,然后根据变量对生物活性影响的重要性进行排序,筛选出前20个对生物活性最具有显著影响的变量[4]。为了便于研究问题,提出几条假设:(1)假设所有数据来源真实可靠,能够反应化合物和分子代表的真实含义[5];(2)假设相关系数矩阵中缺失值的赋值为零,对其他数据没有影响;(3)假设分子描述符之间不存在相互影响,不会发生化学反应;(4)假设数据清洗和数据预处理的步骤均准确无误,也就是能够剔除原始数据中全为0的变量及异常值。
2基于主成分-皮尔逊法对化合物生物活性分子的筛选
2.1研究思路
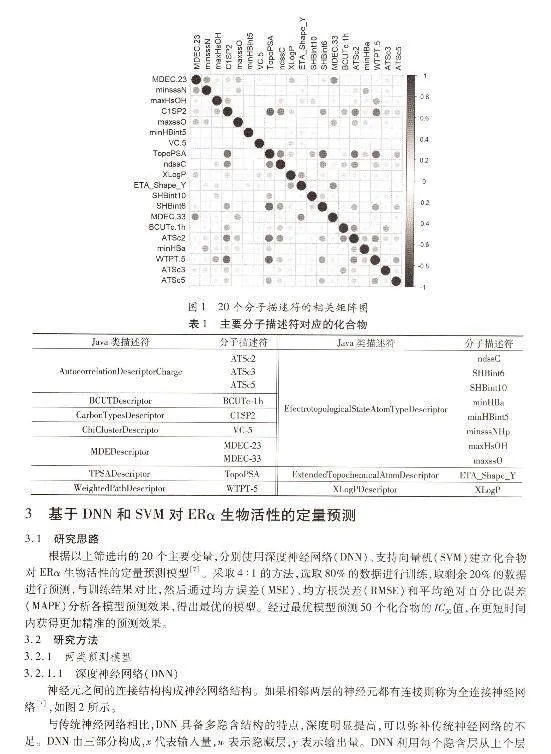
首先建立定量结构-活性关系模型,以分子结构描述符作为自变量,化合物的生物活性值作为因变量,采用主成分分析法降低数据维度[6],按照主成分累计贡献率超过80%确定主成分的个数k,计算主成分得分,确定前100个对生物活性最具有显著影响的分子描述符。考虑到下一步的变量间相关性过滤操作会对进一步变量进行降维,所以在这一步先筛选出100个变量。由于因子贡献率只要达到80%时数据就有效,因此根据这个理论选取前100个变量。然后根据皮尔逊相关系数法算出样本间的相关系数,排除相关性高的分子描述符,保证分子描述符之间的独立性。最后用R语言作出前20个变量的相关矩阵图。
2.2理论准备
主成分分析是考察多个变量间相关性的一种多元统计方法,研究如何通过少数几个主成分来揭示多个变量间的内部结构,即从原始变量中导出少数几个主成分,使它们尽可能多地保留原始变量的信息,此间互不相关。由于数据维数过多会造成模型数据冗长,在建立预测模型前须使用主成分分析法对数据降维。可以将分子描述符的数据分解成能够代替原始数据的低维度数据,提取到对模型最有用的特征值。假设原始分子描述符数据矩阵为A=(X1,X2,…,Xm)(数据的维度为m),先进行中心化:j=aj-1nni=1ai,(1)其中XT=(1,2,…,n),Xi为原始变量,ai为原始变量标准化的数据,=(1,2,…,m),求中心化矩阵为Y=A-,(2)求得协方差矩阵为C=YTY,求得C的m个特征向量按大小顺序组成矩阵W,只取W的前l列求解降维矩阵:T1=YW1,这个低维的分子描述符数据矩阵能够有效减少后续工作量,并有助于模型的特征提取和建模分析。
2.3结果分析
运用主成分分析法对原始数据进行降维处理,综合考虑算法速度和算法准确率,设定k=100,由运行程序得到变量的贡献度排名,并对贡献度由大到小进行排序。主要的问题是选取的100个变量只能保证分子描述与生物活性之间的高相关关系,但是不能保证各个分子描述符之间的独立性,也就是不能排除各个分子描述之间是否存在高相关性。因此,对主成分分析选出的100个变量进行皮尔逊相关系数检验,选出没有相关性或者相关性较低的分子描述符,并且能够提供模型所需的全部信息,由此相关系数保证了自变量之间的独立性。总体来看,主成分分析法和皮尔逊相关系数检验相结合,既保证了自变量与因变量的高度相关,又保证了各个自变量之间是独立的。
通过主成分分析降维和皮尔逊相关系数筛选相关系数低的变量,最终保留20个分子描述符——MDEC-23、minsssN、maxHsOH、C1SP2、maxssO、minHBint5、VC-5、TopoPSA、ndssC、XLogP、ETA_Shape_Y、SHBint10、SHBint6、MDEC-33、BCUTc-1h、ATSc2、minHBa、WTPT-5、ATSc3、ATSc5,它们分别属于10个化合物,详见图1和表1。
3基于DNN和SVM对ERα生物活性的定量预测
3.1研究思路
根据以上筛选出的20个主要变量,分别使用深度神经网络(DNN)、支持向量机(SVM)建立化合物对ERα生物活性的定量预测模型[7]。采取4∶1的方法,选取80%的数据进行训练,取剩余20%的数据进行预测,与训练结果对比,然后通过均方误差(MSE)、均方根误差(RMSE)和平均绝对百分比误差(MAPE)分析各模型预测效果,得出最优的模型。经过最优模型预测50个化合物的IC50值,在更短时间内获得更加精准的预测效果。
3.2研究方法
3.2.1两类预测模型
3.2.1.1深度神经网络(DNN)
神经元之间的连接结构构成神经网络结构。如果相邻两层的神经元都有连接则称为全连接神经网络[7],如图2所示。
与传统神经网络相比,DNN具备多隐含结构的特点,深度明显提高,可以弥补传统神经网络的不足。DNN由三部分构成,x代表输入量,w表示隐藏层,y表示输出量。DNN利用每个隐含层从上个层次得到的向量值,然后进行函数激活的非线性转变,再将向量值传递到下一神经元,一直反复迭代,最后传输到输出层。
3.2.1.2支持向量机(SVM)
SVM作为传统机器学习的一个十分重要的分类算法,自1964年提出以来得到了迅速发展,并发展了一系列改进和扩展的算法,已应用于人脸识别和文本分类等模式识别领域。对于给定训练样本,SVM是一种二分类模型,它的目的是寻找一个超平面来对样本进行分割,分割的原则是间隔最大化,最终转化为一个凸二次规划问题来求解。
SVM模型是找一个分类超平面t=wTx+b,使分类“间隔”最大,即求解二次规划:min12‖w‖2 s.t." yi(wTx+b)≥1,i=1,2,…,n。对于线性不可分的情形,需要加入软间隔及惩罚系数C,分类问题变为如下二次规划:min12‖w‖2+Ckξk s.t. yi(wTx+b)≥1,i=1,2,…,n,ξk≥0。利用拉格朗日乘子法,可转化为max-12klαkαlykyllt;xk,xlgt;+μkαk s.t. kαkyk=0,0≤αk≤C,μ≥0。定义核函数K(xk,xl)=lt;(xk),(xl)gt;代替lt;xk,xlgt;,将上式转化为如下规划问题max-12klαkαlykylK(xk,xl)+μkαk s.t. kαkyk=0,0≤αk≤C,μ≥0,该公式可用于解决非线性分类问题。
3.2.2评价指标
为了评价模型预测结果的好坏,需要一些评价指标对结果进行对比。预测结果评价指标是指采用一系列的指标来评价预测结果的精度,常用的预测结果评价指标是MSE、RMSE、MAPE。
(1)MSE是指均方误差,其数学表达式为MSE=1nni=1(i-yi)2,取值范围为[0,+∞),误差与该值成正比。
(2)RMSE是指均方根误差,即MSE加根号,这样在数量级上比较直观。其计算公式为RMSE=1nni=1(i-yi)2,取值范围为[0,+∞),误差越大,该值也就越大。
(3)MAPE是指平均绝对百分比误差,其数学表达式为MAPE=1nni=1i-yiyi×100%,取值范围为[0,+∞),MAPE大于100%则说明模型较差。但是当真实数据等于0时,存在分母为0的问题,该公式不可用。
3.3结果分析
利用上述的SVM和DNN两种方法来建立ERα生物活性的定量预测模型,两个模型的预测结果与测试结果对比如图3和图4所示。
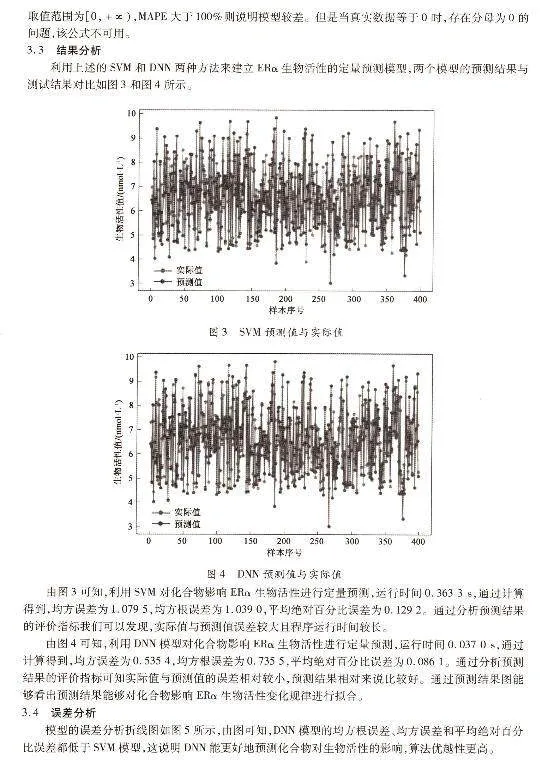
由图3可知,利用SVM对化合物影响ERα生物活性进行定量预测,运行时间0.363 3 s,通过计算得到,均方误差为1.079 5,均方根误差为1.039 0,平均绝对百分比误差为0.129 2。通过分析预测结果的评价指标我们可以发现,实际值与预测值误差较大且程序运行时间较长。
由图4可知,利用DNN模型对化合物影响ERα生物活性进行定量预测,运行时间0.037 0 s,通过计算得到,均方误差为0.535 4,均方根误差为0.735 5,平均绝对百分比误差为0.086 1。通过分析预测结果的评价指标可知实际值与预测值的误差相对较小,预测结果相对来说比较好。通过预测结果图能够看出预测结果能够对化合物影响ERα生物活性变化规律进行拟合。
3.4误差分析
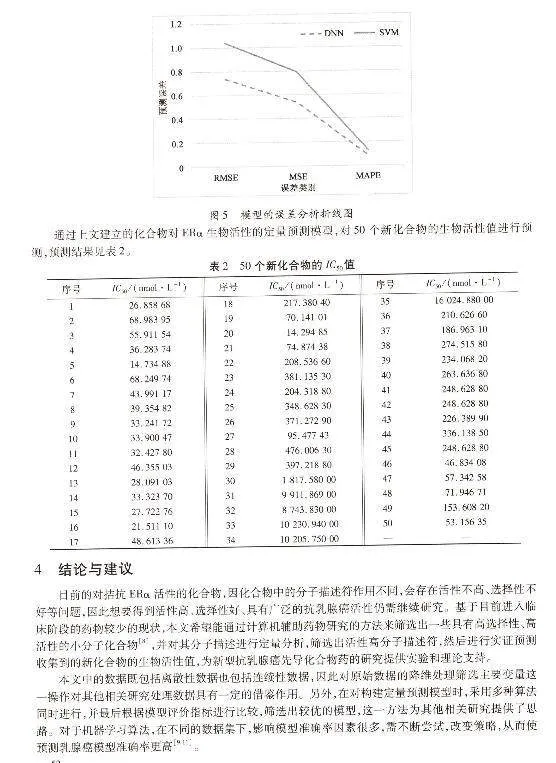
模型的误差分析折线图如图5所示,由图可知,DNN模型的均方根误差、均方误差和平均绝对百分比误差都低于SVM模型,这说明DNN能更好地预测化合物对生物活性的影响,算法优越性更高。
通过上文建立的化合物对ERα生物活性的定量预测模型,对50个新化合物的生物活性值进行预测,预测结果见表2。
4结论与建议
目前的对拮抗ERα活性的化合物,因化合物中的分子描述符作用不同,会存在活性不高、选择性不好等问题,因此想要得到活性高、选择性好、具有广泛的抗乳腺癌活性仍需继续研究。基于目前进入临床阶段的药物较少的现状,本文希望能通过计算机辅助药物研究的方法来筛选出一些具有高选择性、高活性的小分子化合物[8],并对其分子描述进行定量分析,筛选出活性高分子描述符,然后进行实证预测收集到的新化合物的生物活性值,为新型抗乳腺癌先导化合物药的研究提供实验和理论支持。
本文中的数据既包括离散性数据也包括连续性数据,因此对原始数据的降维处理筛选主要变量这一操作对其他相关研究处理数据具有一定的借鉴作用。另外,在对构建定量预测模型时,采用多种算法同时进行,并最后根据模型评价指标进行比较,筛选出较优的模型,这一方法为其他相关研究提供了思路。对于机器学习算法,在不同的数据集下,影响模型准确率因素很多,需不断尝试,改变策略,从而使预测乳腺癌模型准确率更高[9-11]。
[参考文献]
[1]李鸿涛,罗琳,马斌林,等.分子靶向药物在乳腺癌中研究进展[J].肿瘤学杂志,2017,23(3):175-179.
[2]卢皎玲,谢沁沁.基于K-MEANS算法的抗乳腺癌候选药物ERα活性优化研究[J].信息技术与信息化,2021(12):45-48.
[3]吕婷婷,禹文韬,张慧琳,等.面向抗乳腺癌候选药物拮抗雌激素受体α生物活性的定量构效关系模型构建[J].中南药学,2022,20(11):2542-2548.
[4]许美贤,郑琰,李炎举,等.基于PSO-BP神经网络与PSO-SVM的抗乳腺癌药物性质预测[J].南京信息工程大学学报(自然科学版),2023,15(1):51-65
[5]武天琦,耿国强,张相良,等.基于灰色关联度方法的过敏性疾病影响因素分析[J].辽宁工业大学学报(自然科学版),2019,39(6):401-407.
[6]杜雪平.基于机器学习方法的ERα抑制剂活性预测[J].科学技术创新,2022(11):1-4.
[7]王宇轩.基于多尺度卷积神经网络的水果识别技术研究[D].廊坊:北华航天工业学院,2021.
[8]王萍,王蕾,李春艳,等.基于FAK的药物筛选及海洋小分子CY308抗乳腺癌研究[J].中国海洋药物,2021,40(5):23-28.
[9]汪家清,韦哲,张太鹏,等.基于随机森林算法的乳腺癌预测模型的研究[J].中国医学装备,2022,19(1):119-123.
[10]费宇.多元统计分析——基于R[M].北京:中国人民大学出版社,2014:56-78.
[11]何小群.多元统计分析[M].4版.北京:中国人民大学出版社,2015:105-123.
[责任编辑:李 莉]
Prediction of biological activity of breast cancer candidate ERα"based on deep neural network
HUA Rui,ZHU Jia-ming
School of Statistics and Applied Mathematics, Anhui University of Finance and Economics, Bengbu 233041, China
Abstract:In order to study the ERα bioactivity of breast cancer candidate, a quantitative structure-activity relationship (QSAR) model was established by Principal Component Analysis with the biological activity value (IC50) of the compounds as the dependent variable and 729 molecular descriptors as the independent variable. Using the Pearson product-moment Correlation Coefficient, we eliminated the highly correlated variables and came up with the 20 with the most significant effects. The quantitative prediction model of ERα bioactivity was established by using the data package of R language, Support Vector Machine (SVM) and Deep Neural Network (DNN), and the results were compared by using evaluation index. The result of DNN model is good, the root mean square error is 0.735 5, the average absolute error is 0.535 4, and the average absolute error is 0.086 1. The results show that the time and cost of drug development can be greatly saved by computer-aided analysis and prediction of the biological activity of the compounds against ERα.
Key words:breast cancer drugs; ERα bioactivity; principal component analysis; support vector machine; deep neural network; prediction
