
电离辐射调控细胞命运在放射性肺损伤中的作用及治疗进展
2023-04-29 00:00:00顾永清敖兴坤朱娇娇
西安交通大学学报(医学版) 2023年2期

顾永清,军事科学院军事医学研究院辐射医学研究所研究员,博士,博士生导师。全军辐射医学专业委员会秘书长、《西安交通大学学报(医学版)》特邀编委、《国际放射医学核医学杂志》编委。主要从事放射性肺组织损伤与防治关键技术、DNA损伤修复、军事特种环境核辐射健康效应与危害评价等研究。作为项目负责人主持国家自然科学基金7项、军队重点项目分题4项、教育部课题项等10余项,参与国家科技支撑计划、军队重大项目等课题多项。以第一或通讯作者署名在Nucleic Acid Research、Respiratory Research、Genes amp; Diseases等国内外刊物发表论文50余篇,参编专著2部,作为第一完成人获授权国家发明专利2项。第一完成人或主要完成人获省部级科技进步一等奖、省部级科技进步二等奖、省部级医学科技二等奖等科研奖励十余项,获军队优秀专业技术人才二类岗位津贴。
摘要:放射性肺损伤是胸部肿瘤放疗及骨髓移植预处理后常见的并发症,是患者放疗剂量的重要限制因素。一旦放射性肺损伤进展至放射性肺纤维化阶段,将严重降低患者生活质量,同时引起患者呼吸衰竭最终导致死亡。电离辐射可诱导细胞发生凋亡、上皮间质转化、衰老、焦亡及铁死亡等命运,不同细胞响应于电离辐射下的不同损伤形式在放射性肺损伤的发生发展中具有重要作用。本文以电离辐射刺激后不同细胞发生不同命运转归为切入点,简述放射性肺损伤的发病机制,并对其临床防治进行综述。
关键词:电离辐射;放射性肺损伤;放射性肺纤维化;细胞命运
中图分类号:R818
文献标志码:A
DOI:10.7652/jdyxb202302001
Role of cell fate regulation by ionizing radiation in radiation-induced lung injury and its therapeutic progress
GU Yongqing1,2, AO Xingkun1, ZHU Jiaojiao2
(1. Graduate Collaborative Training Base of Academy of Military Sciences,Hengyang Medical School of University of South China, Hengyang 421001;2. Beijing Key Laboratory for Radiobiology, Beijing Institute of Radiation Medicine, Beijing 100850, China)
ABSTRACT: Radiation induced lung injury (RILI) is a common complication after radiation therapy of breast tumors and bone marrow transplantation pretreatment, and it is a critical limiting factor of radiotherapy doses in patients. Once RILI progresses to the radiation-induced pulmonary fibrosis stage, it seriously reduces the patient’s quality of life, while causing the patient’s respiratory failure and eventually leading to death. Ionizing radiation (IR) can induce cell injuries, including apoptosis, epithelial-mesenchymal transition, senescence, pyroptosis and ferroptosis, and these injuries can play an important role in the occurrence and development of radioactive lung injury. Starting from discussion of the occurrence of different forms of injury in different cells after IR stimulation, this review summarizes the pathogenesis of RILI and its clinical prevention and treatment.
KEY WORDS: ionizing radiation; radiation-induced lung injury; radiation-induced pulmonary fibrosis; cell fate
放射性肺损伤 (radiation-induced lung injury, RILI) 是肺癌、食管癌及乳腺癌等胸部肿瘤放射治疗及骨髓移植预处理的常见并发症,极大地限制了肿瘤患者放疗的剂量。临床上将放射性肺损伤分为早期的放射性肺炎 (radiation-induced pneumonitis, RIP) 和晚期的放射性肺纤维化 (radiation-induced pulmonary fibrosis, RIPF) 两个阶段[1-4]。RIP阶段临床常见症状为干咳、呼吸急促及胸痛等;一旦患者发展至RIPF阶段,将出现呼吸困难甚至呼吸衰竭等症状,严重影响患者的生活质量及预后;呼吸衰竭是RILI致死的主要原因,目前临床上针对RILI主要采用对症治疗,尚缺乏高效低毒的防治手段;RILI已经成为放射治疗的重要限制因素。因此,针对RILI的发病机制及防治研究具有十分重要的临床意义。
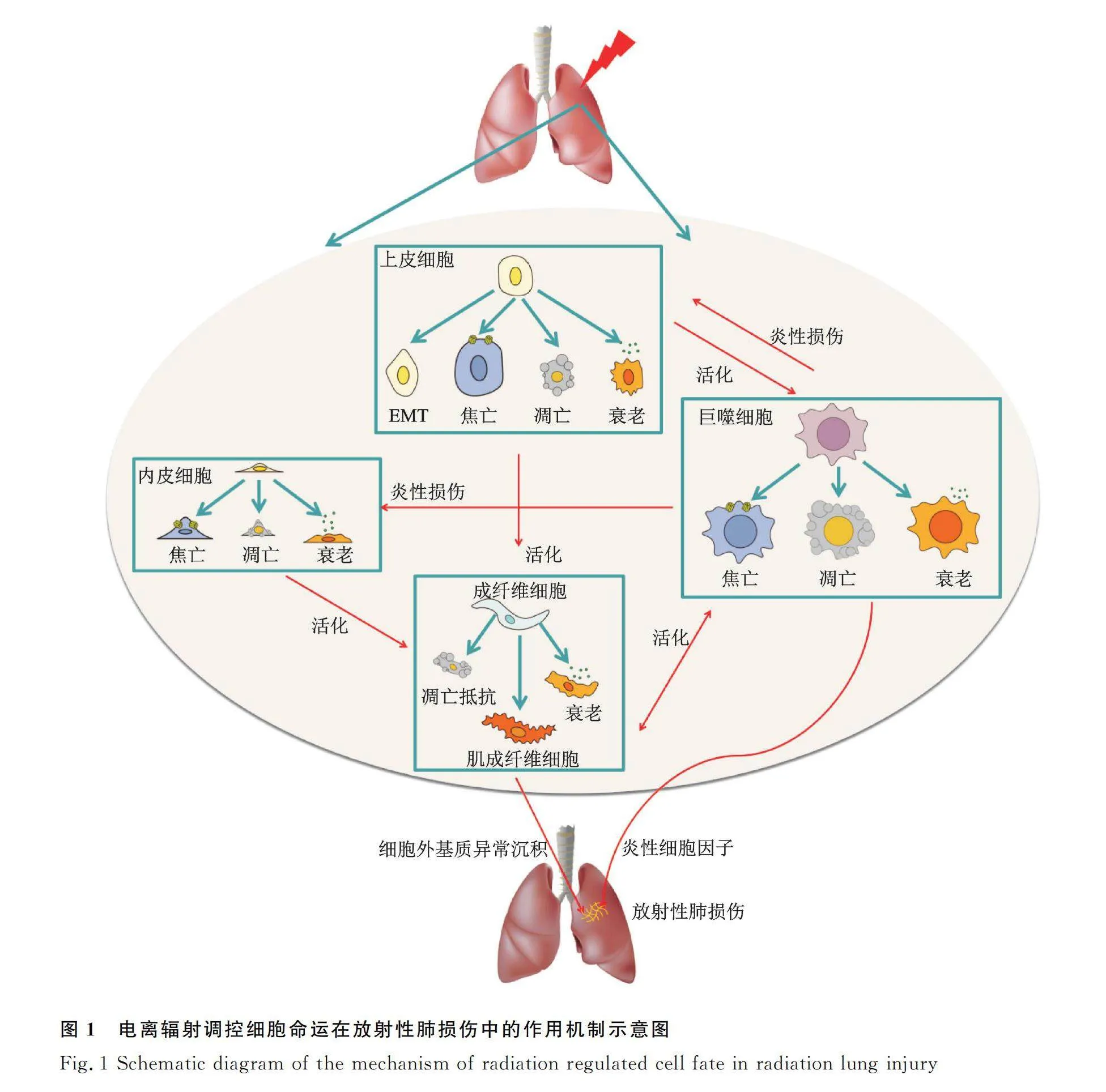
细胞死亡是生物体响应各种刺激因素的一项生命基本过程,包括凋亡、焦亡、铁死亡及铜死亡等。电离辐射刺激下,肺内细胞命运及其调控在RILI发生发展中的作用及机制是值得深入探索的重要科学问题。大量研究证实了RILI是肺泡上皮细胞、肺血管内皮细胞、成纤维细胞及肺泡巨噬细胞等直接或间接响应于电离辐射诱导的凋亡、上皮间质转化 (epithelial-mesenchymal transition, EMT)、衰老、焦亡及铁死亡等多种细胞命运转归形式及多细胞因子共同作用的结果[5-16]。因此,针对电离辐射诱导肺组织内相关细胞损伤及具体机制研究,可能成为RILI防治的新靶点,具有十分重要的临床意义。本文就电离辐射诱导的不同类型肺内细胞命运在RILI中的作用及相关机制进行系统综述归纳,并初步探讨其防治进展。
1 肺内不同类型细胞在放射性肺损伤中的作用
1.1 肺泡上皮细胞
肺泡上皮细胞是放射性肺损伤的关键靶细胞,肺泡Ⅱ型上皮细胞(alveolar epithelial cells Ⅱ, AECⅡ)在肺损伤后可分泌表面活性物质并重建补充Ⅰ型细胞(alveolar epithelial cell Ⅰ, AECⅠ),具有干细胞特性,对维持肺泡组织结构和功能的稳定具有重要作用[17-19]。电离辐射刺激可诱导肺泡上皮细胞损伤,这一方面可导致肺泡上皮细胞耗竭,肺泡结构被破坏;另一方面可释放相应炎性细胞因子、生长因子,加剧肺组织炎症反应[9]。
1.2 内皮细胞
肺内皮细胞亦可响应于电离辐射,直接的内皮损伤导致屏障功能和血管完整性丧失,从而降低了微血管的密度和氧灌注。随着损伤加剧,ROS通过直接激活细胞中的低氧诱导因子来抵抗细胞氧化应激反应,从而导致多种细胞因子和血管内皮生长因子等生长因子被异常激活,促进内皮细胞增殖,这将进一步加剧肺纤维化的形成[20-24]。
1.3 巨噬细胞
传统观念认为肺泡巨噬细胞较少直接响应于电离辐射,其在RILI中的作用主要是被受损的上皮及内皮细胞分泌多种细胞因子激活而诱发“炎症风暴”。但近年来的研究表明,肺泡巨噬细胞亦可直接响应于电离辐射导致细胞衰老及焦亡等,同时还会诱导巨噬细胞极化方向向促炎型进展[25-28]。
1.4 成纤维细胞
成纤维细胞被认为是肺纤维化发生发展过程中最为活跃的细胞,在RILI发展过程中,一方面受损的上皮细胞可通过EMT产生成纤维细胞,另一方面祖成纤维细胞直接响应于电离辐射刺激分泌TGF-β能够通过自分泌机制诱发成纤维细胞的增殖和分化。最终在多种细胞因子的共同作用下,成纤维细胞活化生成肌成纤维细胞,导致细胞外基质异常沉积(extracellular matrix, ECM),加剧RIPF[9,29-32]。
2 电离辐射刺激下,不同细胞命运在放射性肺损伤中的作用
2.1 电离辐射诱导的细胞凋亡
细胞凋亡(apoptosis),也称为程序性细胞死亡(programmed cell death, PCD),是为了维持机体内环境的稳定,在某些因素诱发下启动细胞内相关基因控制的死亡程序,进而引起细胞死亡的过程。细胞凋亡信号通路主要有两条:一条是死亡受体介导的外部细胞凋亡信号通路,通过胞外死亡受体与相应配体结合激活细胞内的凋亡执行分子caspase;另一条是线粒体介导的内部细胞凋亡信号通路,如DNA损伤、缺氧和生长因子缺乏等诱导的凋亡反应,主要通过调控线粒体膜通透性,释放凋亡激活因子来激活caspase[33-37]。在机体胚胎发育、免疫功能的调节及肿瘤发生发展等方面,细胞凋亡均发挥着至关重要的作用。
大量研究证实了电离辐射可导致DNA损伤反应并导致G2/M细胞周期检查点失活,同时通过ROS激活caspase诱导细胞发生明显的G2/M阻滞和S期阻滞,最终导致细胞凋亡[38]。电离辐射诱导的死亡受体Fas高表达呈现一定程度的时间、剂量依赖性,并且与细胞凋亡呈正相关[39-42]。p53作为一种细胞周期调节关键蛋白,其在电离辐射诱导的DNA损伤反应过程中逐渐累积并被磷酸化激活、稳定性增加以促进DNA损伤修复,若DNA不能修复损伤,p53则可以促使高尔基体释放储存的死亡受体,与死亡配体特异结合诱导促凋亡基因Bax表达,导致细胞凋亡增加[43-46]。研究表明,AECⅡ早期直接响应于电离辐射刺激发生凋亡,以清除受损细胞。上皮细胞凋亡将导致上皮细胞耗竭,破坏肺泡结构,并且后期则以其他炎性死亡形式主导,这导致机体无法及时有效清除受损细胞,同时伴随着强烈的炎症反应,加剧RILI[47]。在正常机体中,当肺组织中肌成纤维细胞数目增多会触发细胞凋亡调节系统;但在RIPF患者中,肌成纤维细胞出现凋亡抵抗并且伴随多因素诱导的细胞外基质重构。并且有研究表明,小分子染料IR-780治疗可抑制成纤维细胞分化、促进其凋亡而缓解RIPF[48]。因此,在接受电离辐射后诱导受损上皮细胞尽早凋亡及促进成纤维细胞凋亡可能成为RILI防治的新思路(图1)。
2.2 电离辐射诱导的肺泡Ⅱ型上皮细胞上皮-间质转化
RIPF发病机制复杂,其中最重要的效应细胞是成纤维细胞,而AECⅡ的上皮-间充质转化是RIPF发生发展过程中成纤维细胞的主要来源,这使得研究AECⅡ细胞的EMT过程成为RIPF发病机制研究的热点。EMT是一个上皮细胞获得间质细胞特征的复杂病理过程,伴随着E-cadherin、zo-1上皮细胞标志物减少及Twist、N-cadherin和α-SMA等间质细胞标志物增加,其特点是细胞与细胞间连接缺失及细胞骨架重组,主要发生在组织损伤、伤口愈合、纤维化和肿瘤转移期间。已知的EMT诱发因素包括缺氧、氧化应激、持续性炎症反应及细胞因子表达异常等[8,10,15-16,49]。
肺泡上皮细胞直接响应于电离辐射,AECⅠ凋亡或坏死,AECⅡ通过自我更新和分化为AECⅠ参与肺损伤的修复;当AECⅡ增殖分化过度时,一方面会导致肺泡表面活性剂分泌减少诱发肺不张,另一方面则会促进AECⅡ自身的EMT进程,导致成纤维细胞数目增加。此外,成纤维细胞会在多种细胞因子、生长转化因子诱导下分化为肌成纤维细胞,其具有分泌包括基质金属蛋白酶、胶原蛋白、糖蛋白和纤维连蛋白、层粘连蛋白等细胞外基质的潜能,加剧肺纤维化[50]。INF-γ、TNF-α、TNF-β、IL-1β、IL-6等炎性细胞因子可促进炎性细胞的募集、增殖和分化,改变血管通透性;转化生长因子-β(Transforming growth factor-beta, TGF-β)、PDGF等生长因子可刺激成纤维细胞的趋化运动,TGF-β/Smads信号通路异常激活,通过调节与形成纤维化相关基因Snail、Slug及β-catenin等的表达在纤维化形成中发挥着重要作用[51-53](图1)。
2.3 电离辐射诱导的细胞衰老
细胞衰老(cellular senescence)是指细胞周期处于不可逆持续性阻滞状态,通常分为复制性衰老和早熟性衰老两大类。目前细胞衰老的主要原因通常可归为以下几类:端粒受损、DNA损伤、ROS、持续性炎症刺激、线粒体功能失活及旁分泌诱导作用[54-60]。电离辐射导致细胞受损的本质原因是ROS的生成诱导的持续性氧化应激反应,这将诱导抗凋亡基因的异常表达和细胞代谢异常活跃;此外,电离辐射刺激下,通常伴随着持续性激活的DNA损伤反应(DNA damage response, DDR),DDR可传导到细胞周期关键调控蛋白p53,p53也是电离辐射损伤调控的关键响应分子,通常作为一种DNA损伤修复蛋白,决定着受损细胞的最终结局。其可通过调控NER途径及MMR途径多种关键蛋白表达,参与DNA损伤修复的调节。p53被认为是一个主要的衰老启动因子,在不可修复的损伤中,p53通过转录调控p21,而p21与多种细胞周期蛋白复合物如CyclinD1-Cdk4、CyclinE-Cdk2等结合,参与细胞周期调控诱发细胞周期阻滞,从而使细胞永久性地退出循环,导致细胞衰老[61-62]。同时,衰老细胞可持续性分泌包含细胞因子、趋化因子及基质金属蛋白酶等衰老相关分泌表型(senescence-associated secretory phenotype, SASP),参与成纤维细胞增殖活化、诱导免疫细胞炎症反应、加剧上皮细胞发生EMT及诱发周围细胞旁分泌性衰老,最终加剧RIPF[63-66]。
衰老细胞堆积及分泌SASP可能成为治疗RIPF的新靶点。最近的研究表明,电离辐射刺激下AECⅡ存在持续的细胞衰老,其可能受胰岛素样生长因子-1受体(insulin-like growth factor-1 receptor, IGF-1R)及12-脂氧合酶(12-lipoxygenase, 12-LOX)调节[6,67-68]。AECⅡ细胞衰老一方面可导致肺泡上皮干细胞耗竭,另一方面其释放的SASP因子是RIPF发生发展的重要环节。SASP因子能活化成纤维细胞,保护肌成纤维细胞免于凋亡,促进ECM过度沉积,加剧肺纤维化;同时,SASP能活化巨噬细胞并促进巨噬细胞向促炎M2型极化,使机体免疫系统异常激活进一步产生大量促炎细胞因子,加剧肺部炎症,导致肺纤维化病变;此外,衰老细胞分泌的SASP还可将衰老表型扩散到周围的细胞,引起周围细胞发生旁分泌型衰老。巨噬细胞作为肺内驻留的主要免疫细胞,受电离辐照后,巨噬细胞在体外表现出多种衰老特征[69],包括p16、p21表达增加及SA-β-gal、COX-2、几种促炎细胞因子/趋化因子和氧化应激水平升高。这与小鼠体内结果相一致,这种效应加剧了体内炎症[70]。亦有单细胞测序数据表明,在接受电离辐射刺激后小鼠肺巨噬细胞衰老标志物酪氨酸激酶Fgr高表达,这间接提示了巨噬细胞亦可出现由电离辐射诱导的细胞衰老表型[71]。在受损的肺中,成纤维细胞及肌成纤维细胞通常表现为凋亡抵抗,并且非小细胞肺癌患者接受胸部肿瘤放射治疗后,会出现癌症相关成纤维细胞衰老表型,这促进了肿瘤细胞的增殖及放射抗性。给予衰老细胞清除剂FOXO4-P53干扰肽特异性诱导衰老细胞凋亡之后,一方面增加了肿瘤细胞的放射治疗敏感性,另一方面有效缓解了辐射诱导的肺纤维症状[72],由此证实了电离辐射诱导的成纤维细胞衰老对放射性肺纤维具有一定促进作用(图1)。
2.4 电离辐射诱导的细胞焦亡
细胞焦亡(pyroptosis)是由NLRP1、NLRP3、 NLRC4及AIM2等炎性小体引发的一种细胞程序性死亡,表现为细胞不断胀大直至细胞膜破裂,质膜完整性破坏,导致细胞内容物释放进而引起强烈的炎症反应,其在肺癌、乳腺癌等肿瘤及巨噬细胞介导的炎症反应等疾病中发挥重要作用[73-74]。细胞焦亡的两种途径分别为依赖caspase-1的经典途径和依赖caspase-4/5/11的非经典途径。Caspase-1介导的经典焦亡途径可直接通过炎症小体诱导IL-18、IL-1β等炎症因子表达;而caspase-4/5和caspase-11并不直接参与炎症因子的激活,其通过诱导caspase-1的活化间接促进炎症因子的释放从而引起机体的炎症反应。
最新研究显示,电离辐射刺激下,模式识别受体(pattern recognition receptors, PRRs)激活,促进caspase-1活化,活化的caspase-1一方面切割GSDMD蛋白破坏巨噬细胞膜的完整性,另一方面使IL-1β和IL-18成熟与分泌,进一步募集循环巨噬细胞等炎症细胞向肺内聚集,并诱导M1型巨噬细胞向M2型促炎巨噬细胞极化,产生级联炎症反应,诱导RILI[75-78]。此外,肺内驻留的M2型巨噬细胞作为TGF-β的主要来源,由焦亡诱导的M2型巨噬细胞增加导致肺内活性TGF-β含量增加,从而促进肺成纤维化细胞的增殖、分化及肺细胞的炎症反应,同时导致肌成纤维细胞的数目及活性增加促进肺纤维化的发生,并且已有研究证实了穿心莲内酯可靶向肺巨噬细胞焦亡改善RIPF症状[15,26,79]。除巨噬细胞焦亡外,利用纳米塑料刺激,可通过激活初级支气管上皮细胞NLRP3炎性小体,导致肺泡上皮细胞焦亡来启动肺纤维化进程;并且NLRP3缺失的小鼠可抵抗由博来霉素诱导的小鼠肺纤维化表型。以上研究提示了上皮细胞焦亡可能在RILI过程中也发挥着重要作用[80-81](图1)。
2.5 其他细胞命运形式
电离辐射对人体健康和生命的危害,究其主要根源是放射性或粒子辐射产生活性氧及氧自由基等对机体细胞基因组DNA损伤以及由DNA损伤信号引发的系列细胞学反应。随着科学研究对细胞死亡方式的深入认识,铁死亡(Ferroptosis)是一种由铁依赖的脂质过氧化驱动的细胞死亡形式被研究人员定义,其依赖于铁诱导的氧化应激反应和脂质过氧化反应诱导的细胞毒性。铁死亡细胞表现出非常明显的生物能量和形态特征,包括细胞内的烟酰胺腺嘌呤二核苷酸(NADH)耗尽,但ATP没有耗尽,以及膜完整性的损失,伴随着肿瘤病形态正常细胞核和线粒体减少,线粒体外膜破裂及膜密度增加[82-83]。目前针对铁死亡的机制研究主要集中于以下三个方面:氨基酸和脂质代谢调节、ROS生成调节、铁离子浓度调节[84-87]。自2012年铁死亡被定义以来,其在神经退行性疾病、乙型肝炎、血液系统疾病、肿瘤等机体多种疾病中的作用得到了广泛关注,其在肺组织相关疾病中的研究主要集中于PM2.5这类环境毒物导致的肺损伤、铜绿假单胞菌感染及过敏性哮喘这三类疾病[88-92]。而针对铁死亡在RILI中的作用及机制研究仍然停留在对RILI动物模型给予铁死亡抑制剂治疗可有效缓解由电离辐射诱导的机体肺组织损伤。例如在2019年研究表明,铁死亡抑制剂liproxstain-1可能上调GPX4的蛋白表达,缓解小鼠RILI;仅有一项研究证实了电离辐射可导致肺内皮细胞发生铁死亡,并初步阐明了其作用机制[93-96]。因此,明确铁死亡在RILI中的作用及寻找在RILI过程中可调控细胞铁死亡的靶蛋白对RILI的防治具有十分重要的临床意义。
2022年3月,在国际学术期刊《Science》上定义了一种不同于已知细胞死亡机制的新型细胞死亡方式——铜死亡(cuprotosis),其是通过铜离子与线粒体呼吸中的三羧酸循环中的脂酰化成分直接结合而发生的,导致脂酰化蛋白质聚集和随后的铁硫簇蛋白下调,从而使得蛋白质毒性应激并最终导致细胞死亡[97-98]。机体内铜的摄入、排出以及代谢受调控并维持动态平衡,当体内铜稳态被打破,铜代谢异常或铜诱导的细胞死亡会导致一系列疾病的发生。既往研究证实了乳腺癌、宫颈癌等肿瘤细胞对铜离子具有较正常细胞更高的需求。肝脏作为机体铜离子含量最高的组织,神经系统次之,因此铜离子代谢异常主要导致的疾病集中于肝脏及神经系统。威尔逊病作为机体典型铜代谢异常疾病,其治疗也主要是通过平衡铜离子代谢改善肝功能障碍及神经系统障碍[97]。这提示铜离子代谢在机体发育过程中具有十分重要的作用。最近的研究也表明,在新型冠状病毒肺炎患者血清中铜离子浓度显著增高,且与肺部炎症水平显著正相关,这表明肺组织也可能是铜离子代谢异常的靶组织器官[99]。此外,铜死亡的本质亦是由于线粒体呼吸功能障碍所诱导的,这与电离辐射诱导的细胞损伤本质相一致。因此,铜死亡这一新细胞死亡方式的发现为探索RILI发生发展的机制及防治研究提供了新的研究思路(图1)。
3 放射性肺损伤的防治研究进展
3.1 传统药物治疗
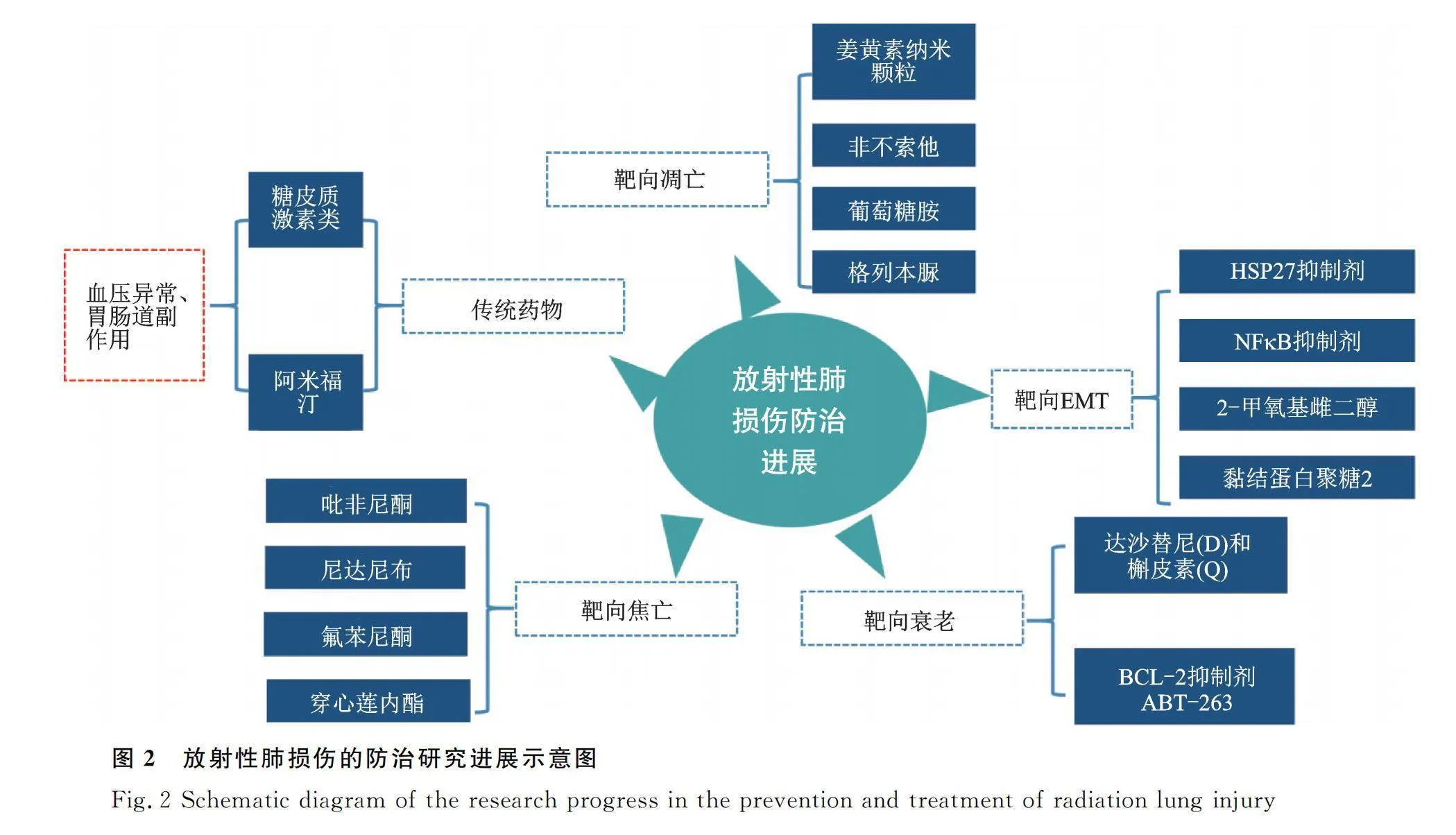
目前针对RILI的临床治疗仍以肾上腺皮质激素治疗为主,而肾上腺皮质激素主要通过减轻血管水肿及炎性渗出,同时伴随着抗生素及祛痰等对症处理,尚缺乏高效低毒的靶向防治手段。长期使用糖皮质激素可诱发骨质疏松、血压升高及钠水潴留等并发症,这极大程度限制了其在防治RILI中的临床应用。同时给予接受电离辐射后的患者细胞保护剂也是一种可用于RILI防治的有效策略。阿米福汀作为首个公认的细胞保护剂,其可通过代谢为能清除电离辐射产生的氧自由基的WR-1065,在临床上也得到了一定的应用。但由于其给药方式单一、代谢时间较快及具有低血压及胃肠道不良反应等副作用,其临床应用也受到了极大的限制[100]。因此,寻找RILI的防治新靶点具有极大的临床应用价值(图2)。
3.2 靶向电离辐射后不同细胞命运治疗放射性肺损伤的研究进展
3.2.1 靶向调节电离辐射诱导的细胞凋亡 近年来的研究发现,靶向细胞凋亡对RILI的治疗具有十分重要的作用。通过制备负载姜黄素的纳米颗粒进行吸入治疗后,可有效抑制电离辐射诱导的肺上皮细胞凋亡,缓解RILI[101]。同时,p53作为关键的促凋亡分子,在电离辐射诱导的细胞凋亡中具有十分重要的作用。研究证实电离辐射可诱导Rac1的核易位,Rac1促进p53的核易位,介导p53依赖性凋亡,抑制Rac1可显著减少正常肺上皮细胞的凋亡,从而有效缓解RILI[102]。非布索他(FBX)是黄嘌呤氧化酶的选择性抑制剂,其可通过抑制caspase-3发挥抗凋亡作用、并且能够缓解肺组织炎症与氧化应激反应,减轻RILI[103]。葡萄糖胺可以抑制电离辐射引起的正常肺上皮细胞的凋亡,促进细胞增殖,以此缓解RILI[104]。格列本脲可通过促进钙离子内流并抑制PKC激活,从而抑制电离辐射诱导的肺血管内皮细胞凋亡,发挥缓解RILI的作用[105]。以上研究表明了靶向电离辐射诱导的细胞凋亡可能成为RILI的有效防治策略(图2)。
3.2.2 靶向抑制电离辐射诱导的肺上皮细胞EMT
靶向EMT在RIPF防治研究中也取得了初步进展。热休克蛋白-27的小分子抑制剂J2(HSP-27)是用于治疗肺纤维化的小鼠模型的候选靶点,在交联HSP-27后,通过抑制IκBa-NFκB信号通路,下调Twist、IL-1β和IL-6的表达来抑制RIPF的发展[106-107]。此外,2-甲氧基雌二醇(2-ME)通过抑制诱导的EMT,逆转电离辐射诱导的HIF-1α水平升高,减少血管胶原沉积,抑制RIPF的发展。这证明了靶向上皮细胞EMT可成为RIPF的有效防治靶点[16,108]。此外,黏结蛋白聚糖2(syndecan-2, SDC2)可以通过抑制PI3K/AKT通路活性,抑制电离辐射刺激下小鼠的成纤维细胞增殖及活化,减轻肺纤维化症状[109-111]。阿米福汀类似物DRDE-30通过减轻炎症和纤维化减轻辐射所致的肺损伤[100]。因此,靶向电离辐射诱导下AECⅡ的EMT过程及EMT后成纤维细胞的活化可能成为RIPF的有效防治手段(图2)。
3.2.3 靶向清除电离辐射诱导的衰老细胞及衰老相关分泌表型 目前,清除衰老细胞及针对SASP干预细胞衰老已成为了研究热点。有研究证实,对具有早衰背景的转基因小鼠敲除p16表达,细胞可以延迟衰老表型的出现或减轻衰老相关症状[112];此外,通过构建人成纤维细胞衰老模型,筛选对SASP具有抑制作用的小分子,发现糖皮质激素及氢化可的松可以减少衰老相关分泌表型的成分,包括几种炎性细胞因子。此外在电离辐射诱导的衰老模型中,糖皮质激素可显著抑制SASP。在许多体外实验和肺纤维化小鼠模型中,激酶抑制剂达沙替尼(D)和类黄酮抑制剂槲皮素(Q)的组合治疗可通过下调p21和p16,促进衰老细胞凋亡,有效治疗肺纤维化[113]。这种治疗效果与p16敲除对肺纤维化的治疗作用相似[114]。同时,BCL-2抑制剂ABT-263亦可有效清除衰老的AECⅡ细胞,有效地降低了衰老的人肺成纤维细胞和小鼠胚胎成纤维细胞的活力[115]。但由于清除药物的靶向性及SASP的高度异质性,使得肺纤维化在内的衰老相关疾病的防治研究仍存在极大的挑战(图2)。
3.2.4 靶向抑制电离辐射诱导的细胞焦亡 吡非尼酮和尼达尼布是已批准用于治疗肺纤维化的药物,可通过抑制NLRP3炎症小体的激活和IL-1β等的分泌,抑制细胞焦亡的发生,减轻肺纤维化及肺部炎症[116];氟苯尼酮是一种结构类似于吡非尼酮的化合物,亦可通过抑制NLRP3炎性小体的激活,降低caspase-1和IL-1β水平,减轻肺纤维化症状[117]。穿心莲内酯是具有抗菌、抗炎和抗癌活性的穿心莲提取成分,其可通过阻止AIM2易位到细胞核以感知辐射诱导的DNA损伤来抑制巨噬细胞焦亡,从而减轻辐射诱导的肺部炎症和纤维化[15]。这表明靶向电离辐射诱导的巨噬细胞及上皮细胞焦亡对RILI的防治具有一定临床价值(图2)。
4 总结与展望
RILI作为限制患者放疗剂量的重要限制因素,其发病机制及防治研究得到了广泛的关注,但其发病机制尚未完全阐明。随着对细胞损伤形式的深入研究,探讨不同细胞损伤形式在RILI中的作用及其成为防治靶点的可行性探索,已经成为RILI的研究前沿,但其中仍有大量空白亟待解决。明确电离辐射刺激下肺组织内各种细胞的损伤在RILI中的作用,阐明其发病机制并在靶向治疗的研发上取得进展性突破,将对RILI的防治具有十分重大的意义。
参考文献:
[1] LI L, WU D, DENG S, et al. NVP-AUY922 alleviates radiation-induced lung injury via inhibition of autophagy-dependent ferroptosis[J]. Cell Death Discov, 2022, 8(1):86.
[2] SHARMA GP, FISH BL, FREI A C, et al. Pharmacological ACE-inhibition mitigates radiation-induced pneumonitis by suppressing ACE-expressing lung myeloid cells[J]. Int J Radiat Oncol Biol Phys, 2022, 113(1):177-191.
[3] ZHANG Z, ZHOU J, VERMA V, et al. Crossed pathways for radiation-induced and immunotherapy-related lung injury[J]. Front Immunol, 2021, 12:774807.
[4] LIU X, SHAO C, FU J. Promising biomarkers of radiation-induced lung injury: A review[J]. Biomedicines, 2021, 9(9):1181.
[5] CHEN JX, YANG L, SUN L, et al. Sirtuin 3 ameliorates lung senescence and improves type Ⅱ alveolar epithelial cell function by enhancing the FoxO3a-dependent antioxidant defense mechanism[J]. Stem Cells Dev, 2021, 30(17):843-855.
[6] CHUNG EJ, KWON S, REEDY JL, et al. IGF-1 receptor signaling regulates type Ⅱ pneumocyte senescence and resulting macrophage polarization in lung fibrosis[J]. Int J Radiat Oncol Biol Phys, 2021, 110(2):526-538.
[7] SU L, DONG Y, WANG Y, et al. Potential role of senescent macrophages in radiation-induced pulmonary fibrosis[J]. Cell Death Dis, 2021, 12(6):527.
[8] YAN Z, AO X, LIANG X, et al. Transcriptional inhibition of miR-486-3p by BCL6 upregulates Snail and induces epithelial-mesenchymal transition during radiation-induced pulmonary fibrosis[J]. Respir Res, 2022, 23(1):104.
[9] MENG J, LI Y, WAN C, et al. Targeting senescence-like fibroblasts radiosensitizes non-small cell lung cancer and reduces radiation-induced pulmonary fibrosis[J]. JCI Insight, 2021, 6(23):e146334.
[10] LIANG X, YAN Z, WANG P, et al. Irradiation activates MZF1 to inhibit miR-541-5p expression and promote epithelial-mesenchymal transition (EMT) in radiation-induced pulmonary fibrosis (RIPF) by upregulating slug[J]. Int J Mol Sci, 2021, 22(21):11309.
[11] WU DM, HE M, ZHAO YY, et al. Increased susceptibility of irradiated mice to Aspergillus fumigatus infection via NLRP3/GSDMD pathway in pulmonary bronchial epithelia[J]. Cell Commun Signal, 2022, 20(1):98.
[12] GUAN D, MI J, CHEN X, et al. Lung endothelial cell-targeted peptide-guided bFGF promotes the regeneration after radiation induced lung injury[J]. Biomaterials, 2018, 184:10-19.
[13] LIU T, YANG Q, ZHENG H, et al. Multifaceted roles of a bioengineered nanoreactor in repressing radiation-induced lung injury[J]. Biomaterials, 2021, 277:121103.
[14] YANG C, SONG C, WANG Y, et al. Re-Du-Ning injection ameliorates radiation-induced pneumonitis and fibrosis by inhibiting AIM2 inflammasome and epithelial-mesenchymal transition[J]. Phytomedicine, 2022, 102:154184.
[15] GAO J, PENG S, SHAN X, et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis[J]. Cell Death Dis, 2019, 10(12):957.
[16] CHOI SH, HONG ZY, NAM JK, et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis[J]. Clin Cancer Res, 2015, 21(16):3716-3726.
[17] WANG LK, WU TJ, HONG JH, et al. Radiation induces pulmonary fibrosis by promoting the fibrogenic differentiation of alveolar stem cells[J]. Stem Cells Int, 2020, 2020:6312053.
[18] LIU X, ZHU X, ZHU G, et al. Effects of different ligands in the notch signaling pathway on the proliferation and transdifferentiation of primary type Ⅱ alveolar epithelial cells[J]. Front Pediatr, 2020, 8:452.
[19] KIM JE, KIM HJ, JUNG JW, et al. TM4SF5-mediated CD44v8-10 splicing variant promotes survival of type Ⅱ alveolar epithelial cells during idiopathic pulmonary fibrosis[J]. Cell Death Dis, 2019, 10(9):645.
[20] KESSLER N, VIEHMANN SF, KROLLMANN C, et al. Monocyte-derived macrophages aggravate pulmonary vasculitis via cGAS/STING/IFN-mediated nucleic acid sensing[J]. J Exp Med, 2022, 219(12):e2022075911022022c.
[21] LAI X, NAJAFI M. Redox interactions in chemo/radiation therapy-induced lung toxicity; mechanisms and therapy perspectives[J]. Curr Drug Targets, 2022, 23(13):1261-1276.
[22] BOUTEN RM, DALGARD CL, SOLTIS AR, et al. Transcriptomic profiling and pathway analysis of cultured human lung microvascular endothelial cells following ionizing radiation exposure[J]. Sci Rep, 2021, 11(1):24214.
[23] BENADJAOUD MA, SOYSOUVANH F, TARLET G, et al. Deciphering the dynamic molecular program of radiation-induced endothelial senescence[J]. Int J Radiat Oncol Biol Phys, 2022, 112(4):975-985.
[24] YANAGIHARA T, TSUBOUCHI K, GHOLIOF M, et al. Connective-tissue growth factor contributes to TGF-β1-induced lung fibrosis[J]. Am J Respir Cell Mol Biol, 2022, 66(3):260-270.
[25] ZHANG Y, ZHU L, HONG J, et al. Extracellular matrix of early pulmonary fibrosis modifies the polarization of alveolar macrophage[J]. Int Immunopharmacol, 2022, 111:109179.
[26] JO WS, KANG S, JEONG SK, et al. Low dose rate radiation regulates M2-like macrophages in an allergic airway inflammation mouse model[J]. Dose Response, 2022, 20(3):15593258221117349.
[27] YAN Y, FU J, KOWALCHUK RO, et al. Exploration of radiation-induced lung injury, from mechanism to treatment: A narrative review[J]. Transl Lung Cancer Res, 2022, 11(2):307-322.
[28] LIU P, LI Y, LI M, et al. Endothelial Shp2 deficiency controls alternative activation of macrophage preventing radiation-induced lung injury through notch signaling[J]. iScience, 2022, 25(3):103867.
[29] SAITO S, DESKIN B, REHAN M, et al. Novel mediators of idiopathic pulmonary fibrosis[J]. Clin Sci (Lond), 2022, 136(16):1229-1240.
[30] SUZUKI T, KROPSKI JA, CHEN J, et al. Thromboxane-prostanoid receptor signaling drives persistent fibroblast activation in pulmonary fibrosis[J]. Am J Respir Crit Care Med, 2022, 206(5):596-607.
[31] CHEN P, LIU H, XIN H, et al. Inhibiting the cytosolic phospholipase A2-arachidonic acid pathway with arachidonyl trifluoromethyl ketone attenuates radiation-induced lung fibrosis[J]. Int J Radiat Oncol Biol Phys, 2023, 115(2):476-489.
[32] DUBEY S, DUBEY PK, UMESHAPPA CS, et al. Inhibition of RUNX1 blocks the differentiation of lung fibroblasts to myofibroblasts[J]. J Cell Physiol, 2022, 237(4):2169-2182.
[33] CHEN H, NING X, JIANG Z. Caspases control antiviral innate immunity[J]. Cell Mol Immunol, 2017, 14(9):736-747.
[34] MAN SM, KANNEGANTI TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity[J]. Nat Rev Immunol, 2016, 16(1):7-21.
[35] BOATRIGHT KM, SALVESEN GS. Mechanisms of caspase activation[J]. Curr Opin Cell Biol, 2003, 15(6):725-731.
[36] KISCHKEL FC, LAWRENCE DA, TINEL A, et al. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8[J]. J Biol Chem, 2001, 276(49):46639-46646.
[37] CHANG D, XING Z, PAN Y, et al. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis[J]. EMBO J, 2002, 21(14):3704-3714.
[38] GANDHI VV, BIHANI SC, PHADNIS PP, et al. Diselenide-derivative of 3-pyridinol targets redox enzymes leading to cell cycle deregulation and apoptosis in A549 cells[J]. Biol Chem, 2022, 403(10):891-905.
[39] CHEN J, LIU X, ZENG Z, et al. Immunomodulation of NK cells by ionizing radiation[J]. Front Oncol, 2020, 10:874.
[40] MONIRUZZAMAN R, REHMAN MU, ZHAO QL, et al. Roles of intracellular and extracellular ROS formation in apoptosis induced by cold atmospheric helium plasma and X-irradiation in the presence of sulfasalazine[J]. Free Radic Biol Med, 2018, 129:537-547.
[41] SATO Y, YOSHINO H, KAZAMA Y, et al. Involvement of caspase-8 in apoptosis enhancement by cotreatment with retinoic acid-inducible gene-I-like receptor agonist and ionizing radiation in human non-small cell lung cancer[J]. Mol Med Rep, 2018, 18(6):5286-5294.
[42] ZHANG J, ZHOU L, NAN Z, et al. Knockdown of c-Myc activates Fas-mediated apoptosis and sensitizes A549 cells to radiation[J]. Oncol Rep, 2017, 38(4):2471-2479.
[43] DENG W, WEI X, XIE Z, et al. Inhibition of PLK3 attenuates tubular epithelial cell apoptosis after renal ischemia-reperfusion injury by blocking the ATM/P53-mediated DNA damage response[J]. Oxid Med Cell Longev, 2022, 2022:4201287.
[44] ZHANG K, WANG L, HONG X, et al. Pulmonary alveolar stem cell senescence, apoptosis, and differentiation by p53-dependent and -independent mechanisms in telomerase-deficient mice[J]. Cells, 2021, 10(11):2892.
[45] SHI T, VAN SOEST D, POLDERMAN P, et al. DNA damage and oxidant stress activate p53 through differential upstream signaling pathways[J]. Free Radic Biol Med, 2021, 172:298-311.
[46] VAN JAARSVELD MTM, DENG D, WIEMER EAC, et al. Tissue-specific Chk1 activation determines apoptosis by regulating the balance of p53 and p21[J]. iScience, 2019, 12:27-40.
[47] SON B, KWON T, LEE S, et al. CYP2E1 regulates the development of radiation-induced pulmonary fibrosis via ER stress- and ROS-dependent mechanisms[J]. Am J Physiol Lung Cell Mol Physiol, 2017, 313(5):L916-L929.
[48] LUO M, CHEN L, ZHENG J, et al. Mitigation of radiation-induced pulmonary fibrosis by small-molecule dye IR-780[J]. Free Radic Biol Med, 2021, 164:417-428.
[49] LEE SY, JEONG EK, JU MK, et al. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation[J]. Mol Cancer, 2017, 16(1):10.
[50] UMBARKAR P, TOUSIF S, SINGH A, et al. Fibroblast GSK-3α promotes fibrosis via RAF-MEK-ERK pathway in the injured heart[J]. Circ Res, 2022, 131(7):620-636.
[51] QU P, SHAO Z, WANG B, et al. MiR-663a inhibits radiation-induced epithelium-to-mesenchymal transition by targeting TGF-β1[J]. Biomed Environ Sci, 2022, 35(5):437-447.
[52] HONG JY, ZAPATA J, BLACKBURN A, et al. A catenin of the plakophilin-subfamily, Pkp3, responds to canonical-Wnt pathway components and signals[J]. Biochem Biophys Res Commun, 2021, 563:31-39.
[53] CHEN Z, GAO H, DONG Z, et al. NRP1 regulates radiation-induced EMT via TGF-β/Smad signaling in lung adenocarcinoma cells[J]. Int J Radiat Biol, 2020, 96(10):1281-1295.
[54] GAMPAWAR P, SCHMIDT R, SCHMIDT H. Telomere length and brain aging: A systematic review and meta-analysis[J]. Ageing Res Rev, 2022, 80:101679.
[55] LIN J, EPEL E. Stress and telomere shortening: Insights from cellular mechanisms[J]. Ageing Res Rev, 2022, 73:101507.
[56] GHADAOUIA S, OLIVIER MA, MARTINEZ A, et al. Homologous recombination-mediated irreversible genome damage underlies telomere-induced senescence[J]. Nucleic Acids Res, 2021, 49(20):11690-11707.
[57] LI X, LI X, XIE C, et al. cGAS guards against chromosome end-to-end fusions during mitosis and facilitates replicative senescence[J]. Protein Cell, 2022, 13(1):47-64.
[58] AGUADO J, SOLA-CARVAJAL A, CANCILA V, et al. Inhibition of DNA damage response at telomeres improves the detrimental phenotypes of Hutchinson-Gilford progeria syndrome[J]. Nat Commun, 2019, 10(1):4990.
[59] JOY J, BARRIO L, SANTOS-TAPIA C, et al. Proteostasis failure and mitochondrial dysfunction leads to aneuploidy-induced senescence[J]. Dev Cell, 2021, 56(14):2043-2058.
[60] YU B, MA J, LI J, et al. Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics[J]. Nat Commun, 2020, 11(1):2549.
[61] STURMLECHNER I, SINE CC, JEGANATHAN KB, et al. Senescent cells limit p53 activity via multiple mechanisms to remain viable[J]. Nat Commun, 2022, 13(1):3722.
[62] FERNANDEZ-DURAN I, QUINTANILLA A, TARRATS N, et al. Cytoplasmic innate immune sensing by the caspase-4 non-canonical inflammasome promotes cellular senescence[J]. Cell Death Differ, 2022, 29(6):1267-1282.
[63] YAO C, GUAN X, CARRARO G, et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis[J]. Am J Respir Crit Care Med, 2021, 203(6):707-717.
[64] ZHANG L, PITCHER LE, YOUSEFZADEH MJ, et al. Cellular senescence: A key therapeutic target in aging and diseases[J]. J Clin Invest, 2022, 132(15):e158450.
[65] HAO X, WANG C, ZHANG R. Chromatin basis of the senescence-associated secretory phenotype[J]. Trends Cell Biol, 2022, 32(6):513-526.
[66] WANG L, CHEN R, LI G, et al. FBW7 mediates senescence and pulmonary fibrosis through telomere uncapping[J]. Cell Metabolism, 2020, 32(5):860-877.
[67] CHUNG E, REEDY J, KWON S, et al. 12-lipoxygenase is a critical mediator of type Ⅱ pneumocyte senescence, macrophage polarization and pulmonary fibrosis after irradiation[J]. Radiat Res, 2019, 192(4):367-379.
[68] CITRIN DE, SHANKAVARAM U, HORTON JA, et al. Role of type Ⅱ pneumocyte senescence in radiation-induced lung fibrosis[J]. J Natl Cancer Inst, 2013, 105(19):1474-1484.
[69] SADHU S, DECKER C, SANSBURY BE, et al. Radiation-induced macrophage senescence impairs resolution programs and drives cardiovascular inflammation[J]. J Immunol, 2021, 207(7):1812-1823.
[70] BERNAL GM, WU L, VOCE DJ, et al. p52 signaling promotes cellular senescence[J]. Cell Biosci, 2022, 12(1):43.
[71] MUKHERJEE A, EPPERLY MW, SHIELDS D, et al. Ionizing irradiation-induced Fgr in senescent cells mediates fibrosis[J]. Cell Death Discov, 2021, 7(1):349.
[72] HAN X, YUAN T, ZHANG J, et al. FOXO4 peptide targets myofibroblast ameliorates bleomycin-induced pulmonary fibrosis in mice through ECM-receptor interaction pathway[J]. J Cell Mol Med, 2022, 26(11):3269-3280.
[73] NIU X, CHEN L, LI Y, et al. Ferroptosis, necroptosis, and pyroptosis in the tumor microenvironment: Perspectives for immunotherapy of SCLC[J]. Semin Cancer Biol, 2022, 86(Pt 3):273-285.
[74] BARRY R, JOHN SW, LICCARDI G, et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity[J]. Nat Commun, 2018, 9(1):3001.
[75] WANG C, YANG T, XIAO J, et al. NLRP3 inflammasome activation triggers gasdermin D-independent inflammation[J]. Sci Immunol, 2021, 6(64):eabj3859.
[76] SONG M, WANG J, SUN Y, et al. Inhibition of gasdermin D-dependent pyroptosis attenuates the progression of silica-induced pulmonary inflammation and fibrosis[J]. Acta Pharm Sin B, 2022, 12(3):1213-1224.
[77] MA L, HAN Z, YIN H, et al. Characterization of cathepsin B in mediating silica nanoparticle-induced macrophage pyroptosis via an NLRP3-dependent manner[J]. J Inflamm Res, 2022, 15:4537-4545.
[78] WU X, YAO J, HU Q, et al. Emodin ameliorates acute pancreatitis-associated lung injury through inhibiting the alveolar macrophages pyroptosis[J]. Front Pharmacol, 2022, 13:873053.
[79] YING H, FANG M, HANG Q, et al. Pirfenidone modulates macrophage polarization and ameliorates radiation-induced lung fibrosis by inhibiting the TGF-β1/Smad3 pathway[J]. J Cell Mol Med, 2021, 25(18):8662-8675.
[80] HUSSAIN S, SANGTIAN S, ANDERSON SM, et al. Inflammasome activation in airway epithelial cells after multi-walled carbon nanotube exposure mediates a profibrotic response in lung fibroblasts[J]. Part Fibre Toxicol, 2014, 11:28.
[81] GASSE P, RITEAU N, CHARRON S, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis[J]. Am J Resp Crit Care, 2009, 179(10):903-913.
[82] WU J, FENG Z, CHEN L, et al. TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models[J]. Nat Commun, 2022, 13(1):676.
[83] LI K, LIN C, LI M, et al. Multienzyme-like reactivity cooperatively impairs glutathione peroxidase 4 and ferroptosis suppressor protein 1 pathways in triple-negative breast cancer for sensitized ferroptosis therapy[J]. ACS Nano, 2022, 16(2):2381-2398.
[84] YAO X, LI W, FANG D, et al. Emerging roles of energy metabolism in ferroptosis regulation of tumor cells[J]. Adv Sci (Weinh), 2021, 8(22):e2100997.
[85] CUI S, SIMMONS G JR, VALE G, et al. FAF1 blocks ferroptosis by inhibiting peroxidation of polyunsaturated fatty acids[J]. Proc Natl Acad Sci USA, 2022, 119(17):e2107189119.
[86] CHEN H, WANG C, LIU Z, et al. Ferroptosis and its multifaceted role in cancer: Mechanisms and therapeutic approach[J]. Antioxidants (Basel), 2022, 11(8):1504.
[87] LI K, XU K, HE Y, et al. Functionalized tumor-targeting nanosheets exhibiting Fe (Ⅱ) overloading and GSH consumption for ferroptosis activation in liver tumor[J]. Small, 2021, 17(40):e2102046.
[88] MAHONEY-SANCHEZ L, BOUCHAOUI H, BOUSSAAD I, et al. Alpha synuclein determines ferroptosis sensitivity in dopaminergic neurons via modulation of ether-phospholipid membrane composition[J]. Cell Rep, 2022, 40(8):111231.
[89] LI J, JIA B, CHENG Y, et al. Targeting molecular mediators of ferroptosis and oxidative stress for neurological disorders[J]. Oxid Med Cell Longev, 2022, 2022:3999083.
[90] BELAIDI AA, MASALDAN S, SOUTHON A, et al. Apolipoprotein E potently inhibits ferroptosis by blocking ferritinophagy[J]. Mol Psychiatry, 2022 . Epub ahead of print.
[91] LI L, WANG K, JIA R, et al. Ferroportin-dependent ferroptosis induced by ellagic acid retards liver fibrosis by impairing the SNARE complexes formation[J]. Redox Biol, 2022, 56:102435.
[92] LI D, PAN J, ZHANG Y, et al. C8orf76 modulates ferroptosis in liver cancer via transcriptionally up-regulating SLC7A11[J]. Cancers (Basel), 2022, 14(14):3410.
[93] YANG Y, MA Y, LI Q, et al. STAT6 inhibits ferroptosis and alleviates acute lung injury via regulating P53/SLC7A11 pathway[J]. Cell Death Dis, 2022, 13(6):530.
[94] GUNES GUNSEL G, CONLON TM, JERIDI A, et al. The arginine methyltransferase PRMT7 promotes extravasation of monocytes resulting in tissue injury in COPD[J]. Nat Commun,2022, 13(1):1303.
[95] LIU X, ZHANG J, XIE W. The role of ferroptosis in acute lung injury[J]. Mol Cell Biochem, 2022, 477(5):1453-1461.
[96] LIU X, WANG L, XING Q, et al. Sevoflurane inhibits ferroptosis: A new mechanism to explain its protective role against lipopolysaccharide-induced acute lung injury[J]. Life Sci, 2021, 275:119391.
[97] PIERSON H, MUCHENDITSI A, KIM BE, et al. The function of ATPase copper transporter ATP7B in intestine[J]. Gastroenterology, 2018, 154(1):168-180.
[98] TSVETKOV P, COY S, PETROVA B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins[J]. Science, 2022, 375 (6586):1254-1261.
[99] IVANOVA I, PAL A, SIMONELLI I, et al. Evaluation of zinc, copper, and Cu:Zn ratio in serum, and their implications in the course of COVID-19[J]. J Trace Elem Med Bio, 2022, 71:126944.
[100] ARORA A, BHURIA V, SINGH S, et al. Amifostine analog, DRDE-30, alleviates radiation induced lung damage by attenuating inflammation and fibrosis[J]. Life Sci, 2022, 298:120518.
[101] CHEN T, ZHUANG B, HUANG Y, et al. Inhaled curcumin mesoporous polydopamine nanoparticles against radiation pneumonitis[J]. Acta Pharm Sin B, 2022, 12 (5):2522-2532.
[102] AN N, LI Z, YAN X, et al. Inhibition of Rac1 attenuates radiation-induced lung injury while suppresses lung tumor in mice[J]. Cell Death Discov, 2022, 8(1):26.
[103] RAEISPOUR M, TALEBPOUR AMIRI F, FARZIPOUR S, et al. Febuxostat, an inhibitor of xanthine oxidase, ameliorates ionizing radiation-induced lung injury by suppressing caspase-3, oxidative stress and NF-kappaB[J]. Drug Chem Toxicol, 2022, 45(6):2586-2593.
[104] LEI X, MA N, LIANG Y, et al. Glucosamine protects against radiation-induced lung injury via inhibition of epithelial-mesenchymal transition[J]. J Cell Mol Med, 2020, 24(18):11018-11023.
[105] XIA P, CAO K, HU X, et al. KATP channel blocker glibenclamide prevents radiation-induced lung injury and inhibits radiation-induced apoptosis of vascular endothelial cells by increased Ca(2+) influx and subsequent PKC activation[J]. Radiat Res, 2020, 193(2):171-185.
[106] KIM JY, AN YM, YOO BR, et al. HSP27 inhibitor attenuates radiation-induced pulmonary inflammation[J]. Sci Rep, 2018, 8(1):4189.
[107] KIM JY, JEON S, YOO YJ, et al. The Hsp27-mediated IkBalpha-NFkappaB signaling axis promotes radiation-induced lung fibrosis[J]. Clin Cancer Res, 2019, 25(17):5364-5375.
[108] NAM J, KIM A, CHOI S, et al. Pharmacologic inhibition of HIF-1α attenuates radiation-induced pulmonary fibrosis in a preclinical image guided radiation therapy[J]. Int J Radiat Oncol Biol Phys, 2021, 109(2):553-566.
[109] TSOYI K, LIANG X, DE ROSSI G, et al. CD148 deficiency in fibroblasts promotes the development of pulmonary fibrosis[J]. Am J Respir Crit Care Med, 2021, 204(3):312-325.
[110] SHI Y, GOCHUICO B, YU G, et al. Syndecan-2 exerts antifibrotic effects by promoting caveolin-1-mediated transforming growth factor-β receptor I internalization and inhibiting transforming growth factor-β1 signaling[J]. AJRCCM, 2013, 188(7):831-841.
[111] RUIZ X, MLAKAR L, YAMAGUCHI Y, et al. Syndecan-2 is a novel target of insulin-like growth factor binding protein-3 and is over-expressed in fibrosis[J]. PloS One, 2012, 7(8):e43049.
[112] LABERGE RM, ZHOU L, SARANTOS MR, et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype[J]. Aging Cell, 2012, 11(4):569-578.
[113] XU M, PIRTSKHALAVA T, FARR JN, et al. Senolytics improve physical function and increase lifespan in old age[J]. Nat Med, 2018, 24(8):1246-1256.
[114] NOVAIS EJ, TRAN VA, JOHNSTON SN, et al. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice[J]. Nat Commun, 2021, 12(1):5213.
[115] PAN J, LI D, XU Y, et al. Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type Ⅱ pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice[J]. Int J Radiat Oncol Biol Phys, 2017, 99(2):353-361.
[116] MAVROGIANNIS E, HAGDORN QAJ, BAZIOTI V, et al. Pirfenidone ameliorates pulmonary arterial pressure and neointimal remodeling in experimental pulmonary arterial hypertension by suppressing NLRP3 inflammasome activation[J]. Pulm Circ, 2022, 12(3):e12101.
[117] SONG C, HE L, ZHANG J, et al. Fluorofenidone attenuates pulmonary inflammation and fibrosis via inhibiting the activation of NALP3 inflammasome and IL-1β/IL-1R1/MyD88/NF-κB pathway[J]. J Cell Mol Med, 2016, 20(11):2064-2077.
(编辑 张 敏)
