
ATP7Bp.Ser1369Tyrfs*24突变致肝豆状核变性1例报告
2023-04-29 00:12:41刘玉婷唐志慧宾琼
临床肝胆病杂志 2023年4期
刘玉婷 唐志慧 宾琼

关键词:肝豆状核变性; 高通量核苷酸序列分析; ATP7B基因
基金项目:广西自然科学基金(2018JJB140029); 广西医疗卫生重点学科项目
A case of hepatolenticular degeneration caused by ATP7B p.Ser1369Tyrfs*24 mutation
LIU Yuting, TANG Zhihui, BIN Qiong. (Department of Pediatrics, The Affiliated Hospital of Guilin Medical University, Guilin, Guangxi 541001, China)
Corresponding author:
BIN Qiong, binqiong1386@126.com (ORCID:0000-0002-0555-4219)
Key words:
Hepatolenticular Degeneration; High-Throughput Nucleotide Sequencing; ATP7B Gene
Research funding:
Natural Science Foundation of Guangxi Province (2018JJB140029); Guangxi Medical and Health Key Disciplines
肝豆状核变性又称Wilson病(Wilsons disease, WD),是由一种铜代谢障碍所致的常染色体隐性遗传病,其发病机制主要是位于第13号染色体长臂(13q14.3~q21.1)上的ATP7B基因突变,导致ATP7B蛋白功能障碍引起铜排泄障碍,进而造成铜大量沉积于组织器官,尤以肝脏、大脑多见[1]。目前WD全球患病率为1/5 000~1/30 000,ATP7B突变基因携带率为1/90[2-3],该疾病主要通过血清铜蓝蛋白、24 h尿铜、青霉胺负荷试验、肝细胞含铜测定及基因测序等检查进行辅助诊断[1]。WD是一种早发现、早诊断、早治疗可有效遏制疾病进展、预后良好的遗传性疾病,因此早期诊断和治疗可大大减少该病的致残率及死亡率。本文通过对先证者及其家系的基因检测并结合相关辅助检查,明确了患儿及其胞兄WD的诊断,进一步丰富了ATP7B基因突变谱。
1 病例资料
先证者,患儿男性,5岁3月龄,1年前因体检发现肝功能异常,未予处理。于2021年11月以“发现肝功能异常1年”为主诉至本院儿科住院。查体:肝肋下4 cm可触及,质韧,边钝,无压痛,余无特殊。完善WD相关检查:肝功能示总胆红素4.95 μmol/L,丙氨酸氨基转移酶180.59 U/L,天门冬氨酸转移酶119.6 U/L;铜蓝蛋白5.9 mg/dL;24 h尿铜92.7 μg;头颅MRI未见明显异常;腹部MRI提示脂肪肝,肝肋下4 cm;眼部检查提示双眼上方角巩膜缘见血管翳,下方角巩膜缘见少量色素沉着,裂隙灯检查未见明显角膜色素环(K-F环);其余检查:凝血功能示纤维蛋白原1.71 g/L;凝血因子检测示多种凝血因子活性降低,其中凝血因子Ⅱ活动度54.5%,凝血因子Ⅸ活动度60.3%,凝血因子Ⅹ活动度65.1%,凝血因子Ⅺ活动度62.9%;肝炎病毒、巨细胞病毒核酸、自身免疫性肝病相关检查均未见异常。追问家族史,先证者胞兄亦有长期肝功能异常病史,检查铜蓝蛋白5.0 mg/dL,父母否认肝功能异常,因此建议该家族完善WD相关基因检测。采集先证者、父母及兄长外周血2~3 mL,根据核酸提取及纯化试剂(博奥木华)操作说明书要求,从外周血中提取基因组DNA。采用QIAseq FX DNA Library Kit试剂盒及定制的单基因病panel,制备包含ATP7B基因全部外顯子及剪切区域的文库,采用AMPure XP beads纯化体系进行纯化并采用Quant-iTTM 1X dsDNA HS Assay Kit和QubitTM荧光计对文库进行定量检测。采用NadPrep NanoBlockers(for Illumina)、NadPrep Hybrid Capture Reagents、KAPA Library Amplification、NovaSeq 6000 Reagent试剂盒,按照操作说明书要求,对先证者文库杂交捕获并进行捕获后产物的扩增、纯化并在NovaSeq 6000测序仪上进行高通量测序。与hg19的参考序列和基因组分析工具包进行比对,进行变异调用。使用ANNOVAR工具对变异进行分析,该工具包含注释数据库,如1000基因组数据库、dbSNP数据库、ClinVar数据库、多态性表型v2数据库以及SIFT数据库。对先证者的可疑致病位点设计相应引物,进行Sanger测序验证。扩增区域1∶chr13∶52523855+52524461,对应的引物序列正向为CAACGTCAAAGTTGACATGATG,反向为TGGATGGGAAAGTCCTGG;扩增区域2∶chr13∶52509545+52510015,对应的引物序列正向为AATTGCCTGCTCATGGTG,反向为CAGCTTTCTAGGAAGCCTCAC,扩增长度为500 bp。采用ABI9700 PCR仪进行扩增,PCR扩增条件如下:95 ℃变性5 min,55 ℃退火30 s,72 ℃延伸30 s,共30个循环,最后72 ℃延伸10 min。产物纯化后用ABI3730基因分析仪进行双向测序,Sequencher软件分析测序结果。
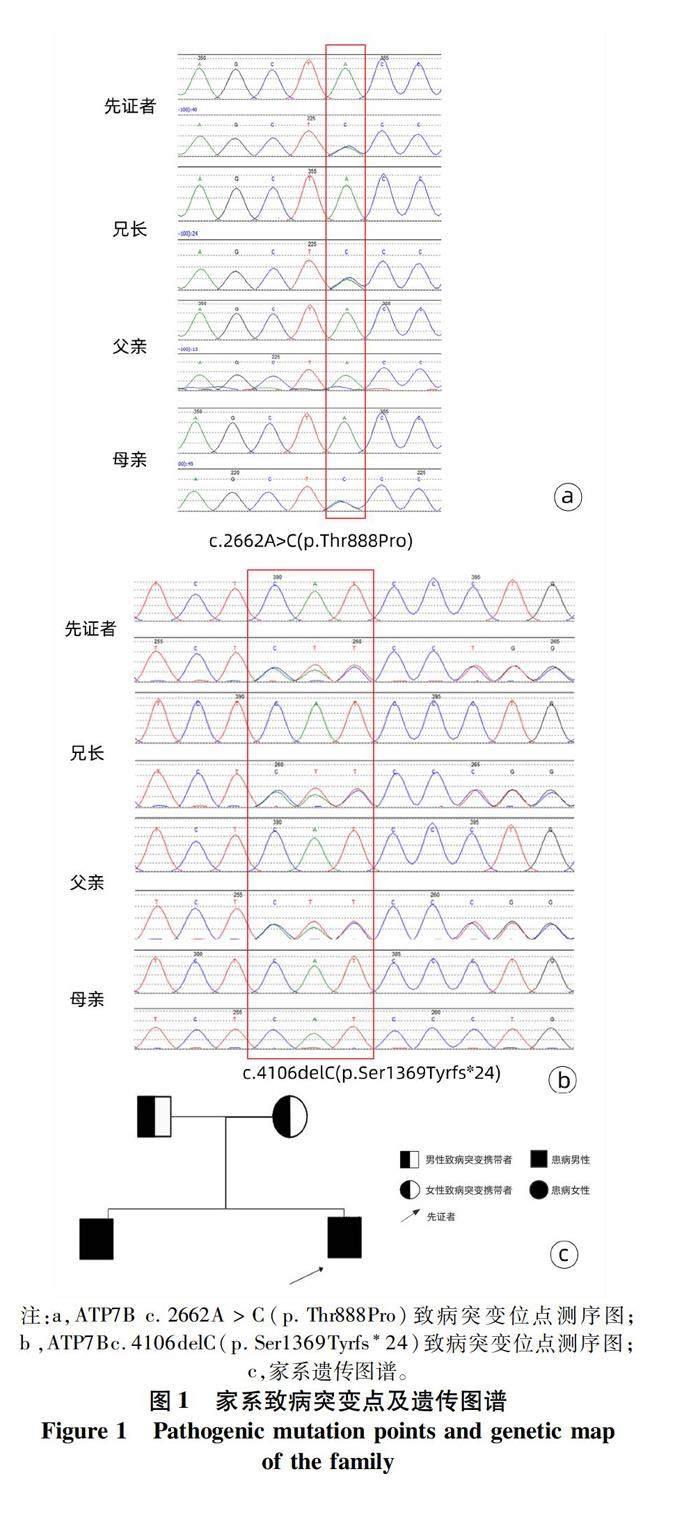
先证者家系遗传图及基因测序结果如图1所示,先证者的ATP7B基因编码区存在2个变异,其中遗传自母亲的c.2662A>C(p.Thr888Pro)为ATP7B基因编码区的错义变异(图1a),该基因位点突变最近已有文献[4-5]报道为WD的致病突变,且与现有文献[6-8]报道的WD致病突变(p.Ile1148Thr;p.Pro992Leu)呈反式排列,故该变异确定为致病变异。另一变异遗传自父亲的c.4106delC(p.Ser1369Tyrfs*24)为ATP7B基因编码区的移码变异(图1b),该变异目前未见ATP7B基因变异数据库和文献报道,为新发突变。同时,先证者兄长的ATP7B基因测序结果同先证者,突变位点详见表1。通过基因分析可见,该家系中父亲的ATP7B基因发生c.4106delC缺失突变,引起第1369位点发生移码突变,由丝氨酸突变为酪氨酸,并继续编码23个氨基酸后终止。此突变理论上导致该基因编码氨基酸序列的提前终止,该变异下游仍有多个无义变异致病的报道[9-11],提示该变异所致氨基酸序列缺失部分对蛋白质功能有重要作用。先证者和兄长均有WD的表型,基因测序结果显示二者基因型相同,根据遗传图谱分析(图1c),父母分别携带1个突变,2个小孩均遗传了2个突变,符合常染色体隐性遗传模式中复合杂合突变致病的机制。根据以上证据,ATP7B c.4106delC(p.Ser1369Tyrfs*24)考虑为新发的致病突变。
2 讨论
WD发病可见于任何年龄,但多起病于儿童及青少年期[12],根据临床症状可分为以下分型。(1)肝型:WD中最常见类型,该型多见于儿童及青少年,以肝损伤为表现,可见进行性转氨酶升高、急性或慢性肝炎、脂肪肝、肝硬化甚至急性肝衰竭,但也可以无症状表现。(2)脑型:以神经系统症状为主,常较肝型发病晚10年,可出现智力倒退、构音障碍、吞咽障碍、运动障碍、不自主运动、震颤、精神症状,该型症状轻时不易发现,察觉时已进入中后期。(3)其他型:如出现肾脏疾病、骨病、肌肉疾病、皮肤色素沉着、K-F环及溶血性贫血等。(4)混合型:以上各型的组合[1,13]。WD的诊断金标准为肝穿刺活组织检查肝细胞铜含量>250 μg/g,但其为有创性检查,故临床应用较少[14]。目前该病主要通过临床表现结合辅助检查如生化指标、影像学及基因检测进行诊断,其中生化指标主要包括血清铜蓝蛋白<200 mg/L、24 h尿铜>100 μg,影像学例如腹部彩超、CT、MRI及头颅MRI(最常见的是基底节区或桥脑的改变)等。结合本病例,先证者为学龄前儿童,以肝脏病变为首发症状,出现转氨酶升高、多种凝血因子活动度降低、脂肪肝,肝大、血清铜蓝蛋白明显降低、尿铜升高等表现,因此考虑可能为肝型WD,为进一步验证,建议家属完善家系基因检测。
随着基因检测技术的不断进步,加之ATP7B基因突变位点的报道增加,基因检测成为WD的一线筛查方法。根据人类基因数据库记载,目前ATP7B基因发现有938个突变位点,种族、地域分布与突变的频率和种类显著相关,其中欧洲WD以c.3207C>A(His1069Gln)为最常见的突变位点,基因频率为13%~61%,亚洲WD以c.2333G>T(Arg778Leu)为常见突变位点,基因频率为34%~38%[13]。另外Cheng等[15]针对中国WD人群研究发现,肝型多见于p.Ser406Ala、p.Val456Leu突变,脑型多见于c.2755C>G(p.Arg919Gly)、c.3443T>C (p.I1e1148Thr)突变,而c.2804C>T(p.Thr935Met)突变常表现为混合型。Li等[16]研究统计结果显示,WD先证者的直系兄弟姐妹患WD的概率为25%,而其子女患WD的概率为0.5%。在欧洲儿童WD诊疗指南[17]中也提出,一旦确诊WD,先证者的一级亲属(兄弟姐妹、后代及父母)均应完善肝功能、铜代谢指标及基因检测。
根据2001年德国莱比锡第8次WD国际会议上制订的Leipzig评分[18],并结合本病例已完善的相关检查结果进行评分,先证者Leipzig评分为5分(24 h尿铜92.7 μg计1分,ATP7B基因外显子测序为复合杂合突变计4分),其兄长Leipzig评分为4分(ATP7B基因外显子测序为复合杂合突变计4分),两者总分均≥4分,故均诊断为WD。
综上,通过本病例研究明确了c.2662A>C(p.Thr888Pro)及c.4106delC(p.Ser1369Tyrfs*24)突变位点为该家系患有WD的遗传因素,同时该家系的c.4106delC(p.Ser1369Tyrfs*24)移码突变为ATP7B基因新发突变位点,进一步丰富了ATP7B基因突变谱。年龄<10岁的患者若以肝脏疾病为首发症状起病时,需警惕WD可能。对高度疑似及确诊的WD患者应進行直系亲属的WD筛查,以便于早发现、早诊断、早治疗,进而有效遏制疾病进展,减少该病的致残率及死亡率。
伦理学声明:本例报告已获得患者家属知情同意。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:刘玉婷负责撰写论文;唐志慧负责收集资料;宾琼负责指导撰写文章,修改文章及最后定稿。
参考文献:
[1]Inherited Metabolic Liver Disease Collaboration Group, Chinese Society of Hepatology, Chinese Medical Association. Guidelines for the diagnosis and treatment of hepatolenticular degeneration (2022 edition)[J]. Chin J Hepatol, 2022, 30(1): 9-20. DOI: 10.3760/cma.j.cn501113-20211217-00603.
中华医学会肝病学分会遗传代谢性肝病协作组. 肝豆状核变性诊疗指南(2022年版)[J]. 中华肝脏病杂志, 2022, 30(1): 9-20. DOI: 10.3760/cma.j.cn501113-20211217-00603.
[2]GITLIN JD. Wilson disease[J]. Gastroenterology, 2003, 125(6): 1868-1877. DOI: 10.1053/j.gastro.2003.05.010.
[3]DONG Y, NI W, CHEN WJ, et al. Spectrum and classification of ATP7B variants in a large cohort of Chinese patients with Wilsons disease guides genetic diagnosis[J]. Theranostics, 2016, 6(5): 638-649. DOI: 10.7150/thno.14596.
[4]LU ZK, CHENG J, LI SM, et al. Phenotypes and ATP7B gene variants in 316 children with Wilson disease[J]. Chin J Pediatr, 2022, 60(4): 317-322. DOI: 10.3760/cma.j.cn112140-20210827-00708.
卢致琨, 程静, 黎丝敏, 等. 肝豆状核变性患儿316例临床表型和ATP7B基因变异特征[J]. 中华儿科杂志, 2022, 60(4): 317-322. DOI: 10.3760/cma.j.cn112140-20210827-00708.
[5]ZHOU J, LIAO JM, LIAO L, et al. A rare ATP7B genotype identified in the siblings with hepatolenticular degeneration and their pedigree analysis[J]. J Clin Hepatol, 2022, 38(5): 1122-1125. DOI: 10.3969/j.issn.1001-5256.2022.05.029.
周洁, 廖金卯, 廖玲, 等. 罕见基因型的肝豆状核变性姐妹及其家系报告[J]. 临床肝胆病杂志, 2022, 38(5): 1122-1125. DOI: 10.3969/j.issn.1001-5256.2022.05.029.
[6]SCHUSHAN M, BHATTACHARJEE A, BEN-TAL N, et al. A structural model of the copper ATPase ATP7B to facilitate analysis of Wilson disease-causing mutations and studies of the transport mechanism[J]. Metallomics, 2012, 4(7): 669-678. DOI: 10.1039/c2mt20025b.
[7]XIAO H, DENG S, DENG X, et al. Mutation analysis of the ATP7B gene in seven Chinese families with Wilsons disease[J]. Digestion, 2019, 99(4): 319-326. DOI: 10.1159/000493314.
[8]HUA R, HUA F, JIAO Y, et al. Mutational analysis of ATP7B in Chinese Wilson disease patients[J]. Am J Transl Res, 2016, 8(6): 2851-2861.
[9]LEPORI MB, LOVICU M, DESSI V, et al. Twenty-four novel mutations in Wilson disease patients of predominantly Italian origin[J]. Genet Test, 2007, 11(3): 328-332. DOI: 10.1089/gte.2007.0015.
[10]BALASHOVA MS, TULUZANOVSKAYA IG, GLOTOV OS, et al. The spectrum of pathogenic variants of the ATP7B gene in Wilson disease in the Russian Federation[J]. J Trace Elem Med Biol, 2020, 59: 126420. DOI: 10.1016/j.jtemb.2019.126420.
[11]ABDELGHAFFAR TY, ELSAYED SM, ELSOBKY E, et al. Mutational analysis of ATP7B gene in Egyptian children with Wilson disease: 12 novel mutations[J]. J Hum Genet, 2008, 53(8): 681. DOI: 10.1007/s10038-008-0298-7.
[12]XIONG F, KUAI Y, XIE SY, et al. The necessity for hepatolenticular degeneration screening among children based on the different results of two cases[J/CD]. Chin J Liver Dis (Electronic Version), 2021, 13(2): 69-72. DOI: 10.3969/j.issn.1674-7380.2021.02.012.
熊复, 蒯钰, 谢双宇, 等. 从2例患儿的不同结局探讨儿童肝豆状核变性筛查的必要性[J/CD]. 中国肝脏病杂志(电子版), 2021, 13(2): 69-72. DOI: 10.3969/j.issn.1674-7380.2021.02.012.
[13]LIANG C, BAI L, ZHENG SJ. Research advances in the genotype-phenotype correlation, diagnosis, treatment, and screening of Wilsons disease[J]. J Clin Hepatol, 2019, 35(9): 2116-2119. DOI: 10.3969/j.issn.1001-5256.2019.09.052.
梁晨, 白麗, 郑素军. Wilson病基因型-表型关系、诊断、治疗及筛查研究进展[J]. 临床肝胆病杂志, 2019, 35(9): 2116-2119. DOI: 10.3969/j.issn.1001-5256.2019.09.052.
[14]OE S, HONMA Y, YABUKI K, et al. Importance of a liver biopsy in the management of Wilson disease[J]. Intern Med, 2020, 59(1): 77-81. DOI: 10.2169/internalmedicine.3440-19.
[15]CHENG N, WANG H, WU W, et al. Spectrum of ATP7B mutations and genotype-phenotype correlation in large-scale Chinese patients with Wilson Disease[J]. Clin Genet, 2017, 92(1): 69-79. DOI: 10.1111/cge.12951.
[16]LI H, LIU L, LI Y, et al. Familial screening of children with Wilson disease: Necessity of screening in previous generation and screening methods[J]. Medicine (Baltimore), 2018, 97(27): e11405. DOI: 10.1097/MD.0000000000011405.
[17]SOCHA P, JANCZYK W, DHAWAN A, et al. Wilsons disease in children: A Position Paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition[J]. J Pediatr Gastroenterol Nutr, 2018, 66(2): 334-344. DOI: 10.1097/MPG.0000000000001787.
[18]BANDMANN O, WEISS KH, KALER SG. Wilsons disease and other neurological copper disorders[J]. Lancet Neurol, 2015, 14(1): 103-113. DOI: 10.1016/S1474-4422(14)70190-5.
收稿日期:
2022-08-04;錄用日期:2022-09-05
本文编辑:葛俊
