
放射性药物的发展状况及监管思考
2023-03-20 02:46:00方笑语
上海医药 2023年5期
方笑语
(上海药品审评核查中心 上海 201210)
放射性药物已经广泛应用于疾病早期发现和炎症组织成像诊断,是分子影像和精准医学的重要基石。随着放射性免疫治疗和放射性多肽类及配体类药物治疗的进步,肿瘤领域的放射性诊疗呈现持续地快速发展,但相关监管政策与技术指导原则尚处于滞后状态。本文对放射性药品研发上市情况、国内放射性药物生产状况及国内外监管法规进行比较分析,探讨该行业目前国内发展主要面临问题并提出监管策略建议。
1 放射性药品研发上市情况
放射性药物按照医疗用途可分为体外放射性诊断试剂、体内放射性诊断用药品、体内放射性治疗用药品。目前国内大多为国外已上市多年品种的仿制药,且各制药公司品种重叠严重,发现新靶点能力不足。特别是为达到精准诊疗目的,近些年提出的靶向放射配体疗法(radioligand therapy,RLT),使得世界各国加强对体内诊断、治疗用特异性配体靶向药物的研发[1-2],这类药品与其他肿瘤治疗方法相比,具有剂量安全,不受常规耐药性机制影响的特点[2],但我国在这方面发展较为滞后,相关放射性药品申报与上市数量有限。
1.1 体外放射性诊断试剂
体外放射性诊断试剂目前常用的包括如碘[125I]系列放射免疫分析药盒、胰岛素放射免疫诊断试剂等。由于近些年在放射性免疫分析(radioimmunoassay,RIA)基础上,逐渐发展起来酶免疫分析、荧光免疫、时间分辨荧光和化学发光等非同位素标记免疫分析技术,使得传统的RIA使用市场逐渐缩小。自上世纪九十年代后,国外RIA所占体外检测份额开始逐年下降,而我国也在2000年后,放射免疫分析药盒生产单位减少到不足10家[3]。
1.2 体内放射性诊断用药品
主要包括单光子放射药物和正电子放射药物。国内上市正电子类仅有用于肿瘤和心脑血管显像的氟[18F]脱氧葡萄糖注射液(18F-FDG),单光子放射药物主要包括锝[99mTc]标记系列药物。而国外,仅配合RLT出现的放射性核素偶联药物(radionuclide drug conjugates,RDC),在近5年就已相继出现上市报道。68Ga-DOTA-TOC作为诊断神经内分泌肿瘤的新型放射性显影剂于2019年成为FDA批准的第1个68Ga标记多肽的放射性药物[4]。2020年12月,FDA批准Telix Pharmaceuticals公司的68Ga-PSMA上市,成为首个获批用于前列腺癌和PSMA阳性病变患者的诊断药物,其研究表明在高危前列腺癌队列中的诊断性能优于磁共振成像[5]。
1.3 体内放射性治疗用药品
目前我国已经批准了2款该类产品进口新药上市,即2020年8月拜耳获得批准的氯化镭[223Ra]注射液和2022年2月远大医药集团之联营公司Sirtex公司的钇[90Y]微球注射液,暂无本土企业新药研发上市。2018年,美国诺华公司的177Lu-DOTA-TATE获得FDA批准上市,这是全球首个获批的多肽受体放射性治疗药物,用于治疗生长抑素受体阳性的胃肠胰腺神经内分泌肿瘤[6]。2021年6月FDA又授予诺华其177Lu-PSMA-617突破性药物资格,用于治疗转移性去势抵抗性前列腺癌[2]。
2 国内放射性药物生产状况
放射性药品的生产工艺一般经过医用核素生产、合成配体、配体标记等步骤。医药核素生产主要通过反应堆辐照、加速器、核素发生器3种方式,而反应堆辐照由于产量大、成本低等优点,是制备医用核素最重要、最常用的方法。由于多种原因,目前我国除少量131I和177Lu外,其他反应堆辐照医用核素均完全依赖于进口,严重阻碍我国放射性药物及核医学的发展。
相对来说,配体的生产厂家在国内众多,供给充足。对配体进行标记,即是对化合物中某一个或多个原子或其化学基团用易辩认的放射性核素进行取代的过程。例如对正电子类放射性药物标记,是在自动合成系统及配套设备组成的全自动卡套式放射性药物标记模块上完成。国产放射性自动化模块多为传统固定管道加电磁阀结构,通常用于11C和18F标记的生产。目前国外已实现68Ga、177Lu等金属核素对小分子、多肽和抗体的全自动放射性标记[7],我国放射性自动化模块已无法满足生产要求。可喜的是,国产全自动卡套式模块设计研究已有报道(图1)[8],但离商业化生产使用还有一段距离。
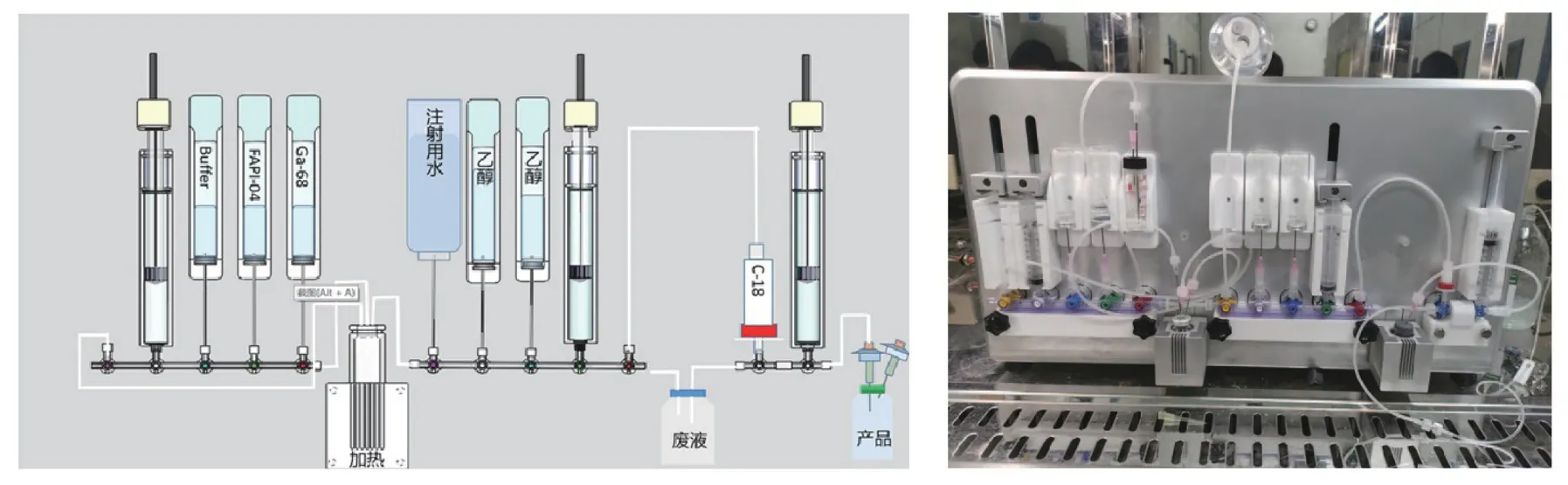
图1 国产68Ga-FAPI-04卡套示意图(左)和Smartlab 20M模块实景图(右)
3 国内外监管法规比较分析
3.1 国外放射性药品相关法规政策和指南
为了更好地监管放射性药品行业发展,国际药品监管机构相继推出或更新了相关的法规及指导原则。2009年,欧盟发布了GMP附录3:放射性药品生产,除了明确GMP相关要求,也指出反应堆、回旋加速器核素生产为非GMP部分。2020年4月,国际原子能机构和WHO发布了“放射性药品GMP指南”正式版,明确了适用场景,同时强调了验证和确认的重要性。国际药品认证合作组织(Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme,PIC/S)于2021年4月发布了GMP附录3,其内容与欧盟完全一致。加拿大卫生部于2020年9月发布了更新后的GMP指南附件3B:正电子发射型放射性药物,用于对这类具有独特的生产、质量控制和处理特点的药品进行额外的指导。随着美国FDA审批流程的改革,一些指导原则也相继出台(表1)。

表1 FDA发布的放射性药物指导原则
3.2 我国放射性药品相关法规政策和指南
2021年6月,国家原子能机构联合科技部等7部门正式发布《医用同位素中长期发展规划(2021—2035年)》,旨在推动医用同位素技术研发和产业发展,重点要求针对国外已应用于临床的放射性诊疗药物,加强技术研发力度,获得一批具备自主知识产权的放射性新药。
我国于2006年发布《医疗机构制备正电子类放射性药品管理规定》,旨在规范医疗机构对正电子放射性药品制备和使用,该规定一直沿用至今。我国也于2012年12月针对放射性药品特点,发布了GMP(2010年修订)的附录六:放射性药品,用于更好地规范企业生产放射性药品行为。为了加强放射性药品的管理,我国在2022年3月根据国务院令752号对1989年国务院颁布的《放射性药品管理办法》进行了第3次修正,将其中企业生产许可、经营许可审批权限下放至省级,强化了省级机构监管责任,进一步推进“放管服”改革,优化营商环境。
为了指导国内放射性药物研究,2020年和2021年我国相继出台了2个技术指导原则,见表2。

表2 国家药品监督管理局药品审评中心发布的放射性药物指导原则
3.3 国内相关法规指南有待完善
针对放射性药物研究,我国仅针对体内诊断药物制定了两个技术指导原则,对体内治疗药物的临床评价与非临床研究还未出台相应的技术指导。近年来国外RLT疗效突出,我国一些品种也处于申请临床试验阶段,急需出台相应的技术指南指导国内企业研究。
在生产方面,监管部门针对放射性药品的特殊性,进行了一些法规实施的细化,如:2022年1月,国家药监局发布关于进一步加强放射性药品管理有关事宜的通告(2022年 第5号),对即时标记放射性药品连续三批样品检验调整至生产企业取得生产许可证后进行,可结合GMP检查动态批同步开展等。但仍存在部分法规实施不适用的现象,如我国GMP(2010年修订)附录六:放射性药品,规定放射性成品留样量应当能够满足注册批准的质量标准2倍的全检量。但根据生产实际情况,生产量较少的药品(如18F-FDG),可能无法达到2倍留样量。对此类情形,欧盟GMP附录3规定单独或少量生产的产品,或其贮存可引起特殊问题的产品的取样或留样,可在获得主管机构同意后,规定其他条件。我国可以参考国外机构根据不同半衰期品种的特点制定灵活的监管要求。对于放射性药品委托检验管理,我国尚无明确的文件规定。一些医疗机构不具备全检条件,是否可以将追溯性检验项目委托第三方,同样没有文件明确规定。
在放射性药品销售管理方面,我国现行药品经营质量管理规范中尚无针对放射性药品经营管理的条款或附录,监督检查中往往以药品GMP产品发运与召回章节中的相关要求作为标准,建议国家药品监管机构借鉴国际上相对成熟的监管经验,结合实际情况,优化放射性药品生产经营管理规范,以推动我国放射性药品高质量发展。
4 目前国内放射性药品发展面临的问题及监管策略的建议
4.1 企业创新力度不足,建议政策支持及发展指引
我国放射性药物企业研发动力不足,市场没被充分挖掘。国内药品监管机构应针对放射性药品新药的研发、申报和审评审批给予一些政策上的宽松与支持。在我国,体内放射性治疗用密封籽源类产品及体外放射性诊断试剂需按照药品注册审批管理,而美国FDA将其划归为医疗器械,由器械和放射产品健康中心下设的体外诊断试剂和放射安全办公室负责管理。与中国最大的不同,FDA对于大部分医疗器械属于事后监管,且密封籽源类产品在《联邦法规典》第21篇892.5730部规定中豁免上市前通告,仅需在FDA系统中完成企业注册和产品列示即可,省去了繁琐的审批流程。
我国放射性药品自主创新性缺乏,同质化比较严重,即便如此,自主设计的生产设备组件商业转化率低。监管机构可举办行业发展供需推介会,加强高校与企业合作。与此同时,监管机构也可建立国际性交流平台,邀请国外放射性药企专家进行行业介绍,如国际上新核素的药物设计应用,有利于加强国内外企业的合作,借鉴国际前沿性研发思路。
4.2 监管条线较多,建议相关部门加强沟通,共建监管方案
放射性药品除本身的药品属性外,它同时具有同位素产品和危险品的属性,因此放射性药品注册过程中,除国家药监局外,还涉及国防科技工业主管部门、国务院环境部等部门审批,各部门监管考虑各自的侧重点,相互支持性不强。
建议药品监管部门牵头,了解企业诉求,积极与其他主管部门沟通,将放射性药品发展中的难点痛点与各方管理要求对接,构建多部门协商合作机制,在达成共识的前提下、不影响药品质量安全的基础上、利用互联网+等技术平台进行结果互认、根据监管风险点制定灵活的审批流程,共建一套科学高效的监管体系,促进新药早日上市。
另外,基于放射性药品需要预约标记配送的特殊要求,建议国家进一步建立统一、科学的放射性药品运输配送细化标准,鼓励引进国内具有危险品运输服务团队及经验资质的大型物流企业涉入配送服务。监管机构可引导企业建立专业化放射性药品配送团队,实现国内资源的高效配置和利用,释放药品生产企业更多活力投身到药物研发和生产活动中。
4.3 专业性监管人才缺乏,建议注重专业队伍建设,科学监管
放射性药品专业性强,但国内高校往往在专业设置上把它归为药学专业的一个分支,很少单独设置,而医学院校中核医学或医学影像学专业更偏重放射性药品临床应用,较少涉及放射性药品的机理研究、质量控制、生产工艺等,因而这方面人才储备少,尤其是专业化的监管人才更是匮乏。
目前国内企业尚未有RDC类药物生产上市销售,这类药物不同于传统放射性药物,主要由介导靶向定位作用的抗体、连接臂、螯合物和放射性同位素构成。生产上如何管控其工艺稳定、质量均一等,是需要国内监管人员思考的。建议药品监管机构与国内外高校进行合作,建立一批监管人才培养基地,开设放射性药品知识相关课程,满足监管人员继续教育需求,注重专业化放射性药品监管人才队伍培养。与此同时,国内药品监管机构可积极争取与国际药品监管机构合作开展人员交流和培训机会,吸取国外对放射性药品企业监管经验。
5 结语
随着国民经济的快速发展,我国的放射性药物市场迅速扩大。但如上文所述,我国自主原创性放射性药物缺乏,医用核素主要依赖于进口,相较于近年来国外法规的更新,我国在放射性药品监管法规方面仍有待完善。本文探讨了目前国内放射性药物发展面临的主要问题,建议相关监管机构对放射性药品企业发展进行政策支持与引导,加强多部门间的沟通,共建监管体系,同时注重专业化队伍建设,进行科学监管,以促进国内该行业发展。
猜你喜欢
中国合理用药探索(2022年1期)2022-11-26 00:22:32
农村青少年科学探究(2022年4期)2022-07-29 14:40:50
中国卫生(2016年5期)2016-11-12 13:25:28
知识经济·中国直销(2016年11期)2016-02-27 16:16:58
环境与生活(2016年6期)2016-02-27 13:46:45
中国卫生(2015年5期)2015-11-08 12:09:48
中国卫生(2015年7期)2015-11-08 11:09:52
太空探索(2015年10期)2015-07-18 10:59:20
中国卫生(2014年7期)2014-11-10 02:33:02
中国卫生(2014年6期)2014-11-10 02:30:42
