
细胞衰老在青光眼中的作用研究进展
2023-03-17 13:24:40丁剑锋综述李璐审校
中华实验眼科杂志 2023年2期
丁剑锋 综述 李璐 审校
1无锡市第八人民医院眼科,无锡 214000;2石河子大学医学院第一附属医院眼科,石河子 832008
青光眼是多种因素引起的以视网膜神经节细胞(retinal ganglion cells,RGCs)丢失为病理改变的不可逆性致盲眼病,眼压升高是青光眼视神经损伤(glaucomatous optic neuropathy,GON)的主要因素。目前,全球青光眼发病率达2.86%,青光眼所致双眼盲人数为1 120万[1];即使能通过药物、激光、手术有效控制眼压,青光眼视神经损害持续进展者仍不鲜见。因此,明确在机械压力、血液动力学异常、免疫炎症、胶质细胞活化等机制中RGCs的损伤进程对青光眼的临床防治尤为重要[2]。房水动力学异常与高眼压密切相关,房水外流通道结构和功能异常参与青光眼的发生和发展。研究认为,细胞僵硬度增大、线粒体功能障碍、蛋白酶体活性下降、胞内信号通路异常等导致小梁网细胞和Schlemm管内皮细胞衰老、通透性及机械传感性降低是青光眼眼压升高的直接危险因素[3]。那么,进一步阐明细胞衰老机制,从而有效抑制房水流出通道无疑是青光眼眼压调控的新策略。细胞生理功能衰退、胞内信号传递及物质运输障碍、DNA损伤等导致细胞周期停滞、细胞发生衰老,在神经退行性疾病及心血管疾病中具有负面推动作用。作为细胞死亡的前一阶段,不同类型细胞发生衰老的机制有独特性,也有共同性[4]。本文就细胞衰老特征、诱因、分子信号通路及其在青光眼中的研究进展进行综述。
1 细胞衰老特征与标志物
细胞衰老发生于所有类型的细胞。有丝分裂细胞中由于DNA损伤反应、细胞周期依赖蛋白激酶(cyclin-dependent kinase,CDK)抑制因子增加等表现出复制性衰老特征,具有再生能力的组织细胞减少、萎缩和损伤修复障碍[5]。RGCs等终末分化细胞中因合成、代谢障碍表现为非复制性细胞衰老,细胞特定功能,如神经信号传递、能量转换等生理功能丧失[6]。衰老细胞结构上发生体积增大、核染色质和线粒体减少、核凹陷等;功能上表现为细胞周期停滞、复制能力丢失、G0期神经元细胞周期异常进入等。目前,应用的细胞衰老标志包括衰老相关-β-半乳糖苷酶(senescence-associated β-galactosidase,SA-β-gal)活性升高、细胞周期调控因子高表达、端粒缩短、衰老相关易染色质聚集(senescence-associated heterochromatic foci,SAHF)和衰老相关分泌表型(senescence associated secretory phenotype,SASP)产生等[7-8]。
1.1 SA-β-gal
SA-β-gal是目前公认的、广泛应用的细胞衰老标志物。病理损伤刺激下,pH 6.0衰老细胞SA-β-gal呈现高酶活性,SA-β-gal作用于神经节苷脂、糖蛋白等底物并将其转变成蓝色,因染色简单、重复性好、普通显微镜下即可检测而广泛应用于细胞衰老研究[9]。Skowronska-Krawczyk等[10]在急性高眼压模型鼠视网膜铺片中应用RGCs标志物Brn3a与SA-β-gal共标,发现高眼压诱导RGCs衰老增加。但是,SA-β-gal染色存在一定局限性,在溶酶体活动旺盛的巨噬细胞、DNA受损细胞中SA-β-gal可能出现假阳性[11]。
1.2 细胞周期调控因子
P16INK4a蛋白又名细胞周期依赖性蛋白激酶抑制因子2A,由INK4a/ARF基因编码,是常用的、可在体检测细胞衰老的标志物。通常,P16INK4a蛋白在正常组织中表达水平极低;炎症、氧化应激等刺激因素导致P16INK4a表达上调引起细胞衰老;因此,P16INK4a高表达也被视为细胞衰老的始动因素[12-13]。
P21Cip1蛋白又名细胞周期依赖性蛋白激酶抑制因子1A,是细胞衰老相关基因P53的下游因子,在小鼠神经管、内淋巴囊等非分裂细胞中检测出P21Cip1与SA-β-gal的高表达[14],在视网膜RGCs损伤中尚未见相关报道。
1.3 SAHF
SAHF是Narita等[15]在成纤维细胞中观察到衰老细胞特有的现象。激光扫描共聚焦显微镜下,衰老细胞核中DNA点状聚集;K9M-H3蛋白、HP1蛋白、HMGA蛋白和组蛋白H2A变异体等标志性蛋白组成SAHF[15],是细胞衰老的重要标志之一。然而,SAHF也存在于部分DNA损伤的细胞中,标记衰老细胞具有一定局限性。
1.4 SASP
SASP是衰老细胞分泌功能增强的重要特征,普遍存在于体内不同形式衰老细胞中。衰老细胞增多、促炎因子、趋化因子、不可溶蛋白、多种蛋白酶等SASP过度累积表现出负性调节作用,参与肿瘤血管生成、慢性炎症、年龄相关性疾病发生等。Coppé等[16]认为不同因素刺激的不同细胞中SASP成分有区别,炎性因子白细胞介素(interleukin,IL)-6/IL-8在SASP中表达较为普遍;而基质金属蛋白酶活化影响小梁网细胞外基质代谢失衡,进而引起房水外流阻力改变是青光眼发生的病理因素。作为反映细胞衰老的指标,合理干预SASP可以为衰老相关糖尿病、阿尔茨海默症、青光眼等疾病的防治提供新的思路。
2 细胞衰老诱因
Hernandez-Segura等[6]研究发现,面临外界应激压力时,细胞正常生理功能和增生能力衰退,进而脱离细胞周期表现为细胞衰老。已有研究证实,紫外线照射、电离辐射、DNA损伤等引起细胞端粒缩短、DNA片段缺失、SAHF聚集,是导致细胞周期停滞、细胞衰老的诱因。致癌基因c-Myc、H-Ras等激活刺激丝裂原活化、产生异常增生信号,进而参与P53、P16信号通路,诱导细胞衰老发生[12,15]。“氧自由基学说”认为,活性氧(reactive oxygen species,ROS)过量产生导致线粒体膜流动性减少、ATP合成减少,胞内衰老信号通路激活,细胞发生线粒体依赖的复制性衰老[17]。
3 细胞衰老经典信号通路
3.1 P16INK4a/Rb途径
P16INK4a/Rb途径是细胞发生衰老的主要途径。细胞受到氧化应激、炎症刺激、电离辐射等损伤引起P16INK4a高表达,竞争性结合CDK4/6进而抑制底物Rb磷酸化,下游转录因子E2F无法被激活使细胞停滞于G1期(图1),引发细胞衰老并表现出SAHF、细胞僵硬度增大、线粒体功能障碍等特征。研究发现,转录因子JunB、JMJD3、HBP1、SP1、TEAD3/4等能正性调控P16INK4a,而ZBP-89、YB1、BMi-1等转录因子则具有负性调控作用[18-20];如何抑制P16INK4a过表达、减缓端粒缩短是目前延缓细胞衰老、增强组织损伤修复能力的重要靶点。
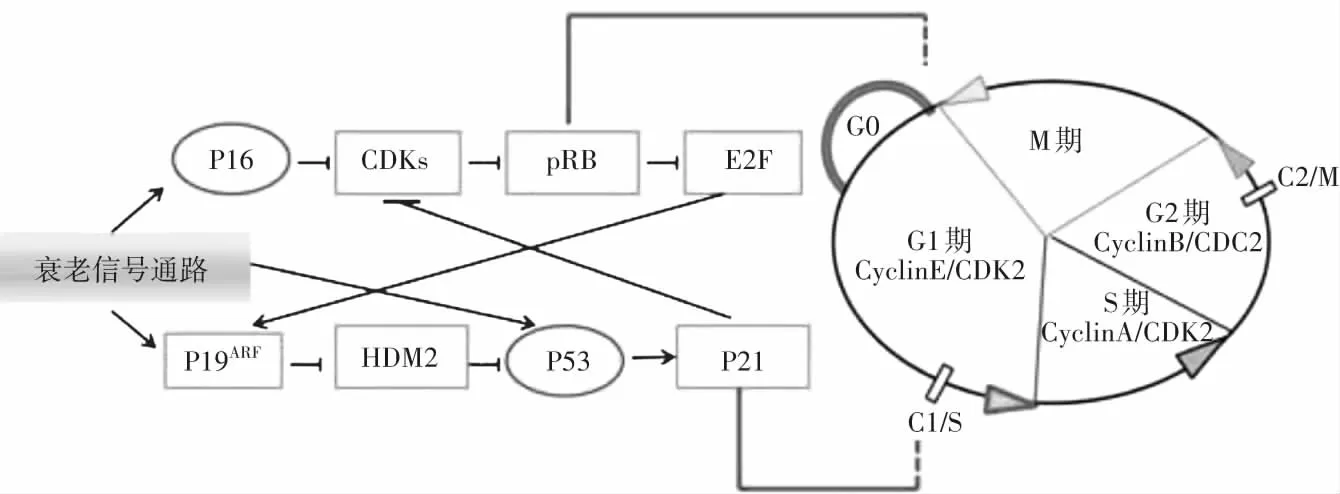
图1 细胞衰老P16INK4a/Rb和P19ARF/P53/P21Cip1途径触发细胞周期阻滞示意图[14] ROS、DNA损伤等刺激信号激活细胞衰老途径P16INK4a/Rb、P19ARF/P53/P21Cip1,衰老细胞中内源性抑制物P21、P16等被激活,引起细胞周期阻滞
3.2 P19ARF/P53/P21Cip1途径
P19ARF/P53/P21Cip1和P16INK4a/Rb衰老信号通路既相互独立,又相互作用,共同参与人类细胞衰老的调控(图1)。生理条件下,INK4a/ARF基因编码P19ARF结合并抑制泛素蛋白连接酶人双微体2蛋白(human double minute 2,HDM2)过表达,进而抑制P53活化。病理条件下,P19ARF蛋白高表达抑制HDM2活性、激活P53,诱导下游因子P21CiP1表达,进而抑制CDK4/6参与的Rb磷酸化激活,细胞周期停滞于G1期,发生细胞衰老[21-22]。作为细胞死亡前一阶段,细胞衰老标志物和机制研究可以为疾病的早期临床防治提供新的思路。
4 青光眼相关的细胞衰老
4.1 青光眼房水引流通路中细胞衰老研究
病理性眼压升高与原发性开角型青光眼(primary open-angle glaucoma,POAG)发生和进展密切相关。眼压稳定有赖于房水生成与流出的动态平衡,睫状突房水生成、房水流出通道阻力以及上巩膜静脉压的变化均能影响眼压稳定[23]。其中,房水流出通道阻力研究是关注热点,也是目前POAG微创手术开展的理论基础;然而何种分子机制参与房水流出通道阻力增加至今尚待阐明。生理状态下,大部分房水经葡萄膜小梁网、角巩膜小梁和邻管组织、Schlemm管内壁内皮细胞微孔进入Schlemm管腔,经过集合管开口流入集合管、房水静脉,最终经表层巩膜静脉代谢。形态学检查发现,房角附近纤维颗粒物质沉积、小梁网细胞分泌的细胞外基质聚集、小梁细胞SA-β-gal染色阳性呈现衰老表型等因素导致筛网状结构破坏、房水流出阻力增加[24-26]。随年龄增长,频域光相干断层扫描检测到Schlemm管径和面积减小[27];此外,Schlemm管内皮细胞衰老呈现出数量和空泡形成减少,也是导致房水外流阻力增大的不可忽视因素[26]。
目前,对小梁网结构和细胞功能的研究主要借助于分离人眼小梁细胞、传代培养猪小梁细胞等,实验中发现Ⅵ型胶原蛋白、纤连蛋白、糖蛋白、糖胺多糖等增加促进小梁网成纤维细胞内衰老标志物SA-β-gal活性增强、有丝分裂减少、细胞形态上皮化、细胞僵硬度增加,提示小梁网细胞衰老与房水流出阻力增加有关[28-29]。电子显微镜下观察发现,紫外线照射、氧化损伤、机械损伤等引起小梁网细胞线粒体中ROS含量增多、ATP合成水平下降和功能障碍[29-30]。研究证实,微小RNA(microRNA,miR)-204异常调控影响线粒体蛋白表达,参与线粒体依赖的复制性细胞衰老发生[30]。此外,有研究发现,氧化应激损伤、miR-106拮抗剂能诱导人小梁细胞和成纤维细胞中P53高表达,进而引起细胞衰老。转染P53小干扰RNA或miR-106激动剂可以有效抑制P53下游因子P21的表达,减少细胞衰老,提示P53/P21Cip1途径参与介导小梁网细胞衰老、房水外流阻力增加[31]。miR-29b表达减少和房水中成纤维细胞生长因子、转化生长因子、胰岛素样生长因子在缺血缺氧、炎症刺激因素下激活,引起小梁细胞外基质合成增加、分泌卷曲相关蛋白1表达上调,导致细胞衰老、僵硬度增加、小梁网通透性破坏,进而引起房水外流受阻增加[32-33]。青光眼被认为是与衰老相关的疾病,蛋白酶体活性下降、P2Y受体激活引起小梁网细胞内Ca2+浓度增加、自噬功能障碍影响细胞机械传感性受损等也与小梁网功能衰退有关[34],但是否存在P16INK4a/Rb途径活化、SAHF聚集等尚需深入研究。
小梁网和Schlemm管是房水外流阻力产生的主要部位。正常情况下,75%的阻力来自于Schlemm管0~14 μm处。Schlemm管内壁内皮细胞基底膜不完整、管腔长度缩短、塌陷引起房水外流受阻;并且,Schlemm管内皮细胞功能衰退呈现出机械传感性、通透性和滤过率下降等,被认为与POAG患者Schlemm管房水外流阻力增加关系密切[35-36]。研究者曾采用手术缝线置入Schlemm管获取人眼Schlemm管内皮细胞,并以CD31分离纯化,由于培养传代困难,目前研究仅限于间接探讨Schlemm管内皮细胞屏障功能和分子机制[37]。此外,有研究在猴、鼠等青光眼动物模型中观察到Schlemm管内皮细胞骨架蛋白、黏蛋白及紧密连接蛋白表达上调;内皮型一氧化氮合酶活性降低导致NO表达减少等机制参与Schlemm管细胞衰老发生[38]。近年来,有研究者关注到Schlemm管细胞骨架结构在青光眼损伤刺激因素作用下的变化,Schlemm管细胞骨架变化是否与Schlemm管内皮细胞衰老有关,目前尚未见报道[39]。鉴于Schlemm管内皮细胞分离困难,Schlemm管内皮细胞衰老在青光眼房水动力异常中的作用尚需深入研究,如何早期发现房水流出通道阻力异常是青光眼早期防治亟待解决的核心问题。
4.2 GON中细胞衰老的研究
RGCs损伤是视功能不可逆丧失的根本原因。病理性眼压升高是始动因素,随后炎症、氧化应激、免疫介导的神经胶质细胞活化、谷氨酸受体NMDA释放、ROS沉积、热休克蛋白及自噬相关蛋白OPTN激活等共同参与GON[40]。现阶段,有效控制眼压联合神经营养因子、扩血管药物支持治疗等手段的综合应用,旨在最大程度地延缓视功能损伤、维持患者生存质量;因此,探索RGCs损伤机制,从而更早、更有效地保护RGCs是青光眼研究者关注的热点问题。
视网膜是中枢神经系统的外延,RGCs被认为是终末分化细胞,始终处于细胞周期G0期;随着中枢神经系统疾病研究的不断深入,病理情况下神经元会再次进入细胞周期,细胞周期再激活表明神经元走向衰老、死亡[14]。青光眼是与阿尔茨海默病、帕金森病在发病机制中有相关的神经退行性疾病,处于G0期的RGCs是否存在异常进入细胞周期的情况以及RGCs结局如何尚待进一步研究。对于RGCs衰老的研究最早始于1986年,Katz等[41]检测到小鼠衰老过程中视网膜外核层、内核层、RGCs层细胞密度显著减少。近年来,有研究者在发育小鼠研究中发现视网膜双极细胞衰老,同时也观察到RGCs中SA-β-gal活性升高[42];Skowronska-Krawczyk等[10]在急性高眼压诱导的视网膜缺血-再灌注损伤动物模型中检测到RGCs衰老,并阐明青光眼风险基因SIX6介导P16INK4a高表达引起RGCs衰老的分子机制。国内也有研究证实,急性高眼压模型鼠中TBK1能促进P16INK4a表达上调,与RGCs衰老有关,并且发现细胞周期S期标志物CyclinA高表达,这些研究结果提示青光眼高眼压损伤作用下神经细胞周期异常参与RGCs衰老进程[43]。此外,有研究发现,OPTNE50K变异导致线粒体自噬和功能障碍,进而参与POAG患者RGCs衰老、胶质细胞活化[44]。RGCs衰老作为青光眼视功能损伤早期阶段,P19ARF/P53/P21Cip1信号通路是否参与缺血缺氧、氧化应激、炎症等刺激因素诱发的RGCs早期损伤还需进一步研究;如何早期在体识别RGCs衰老也是青光眼视功能损伤早期防治研究的方向。
修复受损神经、改善神经信号传递、早期防治RGCs衰老是延缓青光眼视功能损伤的新策略。Rocha等[45]发现达沙替尼可以清除衰老RGCs、抑制SASP,从而保护RGCs和延缓视功能损伤、减轻GON。Huang等[46]研究发现,CYP2J2能够抑制青光眼高眼压损伤引起的血管内皮细胞衰老标志物P16、P53高表达,对视网膜血管和RGCs有保护作用。此外,有实验证实,维生素B3可以提高视网膜烟酰胺腺嘌呤二核苷酸水平、调控线粒体功能,从而延缓青光眼RGCs衰老;艾地苯醌、亚甲蓝可以抑制青光眼氧化应激损伤因素下细胞色素C释放、RGCs中SA-β-gal活性升高[47-48]。培养神经干细胞替换神经系统中衰老细胞仍处于探索阶段,如何干预靶基因、药物治疗如何向临床转化是延缓视功能损伤、青光眼早期防治的重点。
综上所述,青光眼发生和发展是多种致病因素、多种机制共同作用的结果。细胞衰老是细胞死亡的前一阶段,在青光眼发生和视功能损伤中的作用机制研究集中在2个方面:小梁网及Schlemm管内皮细胞衰老、通透性下降,房水流出阻力增加参与青光眼发生;识别和干预RGCs衰老是青光眼视功能早期防治的新策略。在体、精准检测出细胞衰老并实现细胞衰老干预的临床转化是青光眼研究新的切入点,也是青光眼视功能保护工作中迫在眉睫的核心问题。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
音乐教育与创作(2020年9期)2021-01-04 01:01:30
国际呼吸杂志(2019年4期)2019-03-12 01:07:30
今古传奇·故事版(2017年23期)2018-01-12 20:20:13
湖南师范大学学报(医学版)(2016年3期)2016-08-03 08:34:20
国际眼科杂志(2016年5期)2016-05-17 03:32:16
中华老年多器官疾病杂志(2016年7期)2016-04-28 08:43:05
癌症进展(2016年10期)2016-03-20 13:15:43
国际眼科杂志(2016年6期)2016-03-10 10:13:32
中国卫生标准管理(2015年10期)2016-01-15 00:48:56
医学研究杂志(2015年5期)2015-06-10 06:43:26
