
Selenized liposomes with ameliorative stability that achieve sustained release of emodin but fail in bioavailability
2023-03-14 06:52MujunZhuShipingZhuQiuoLiuYuehongRenZhiguoXingwngZhng
Chinese Chemical Letters 2023年1期
Mujun Zhu,Shiping Zhu,Qiuo Liu,Yuehong Ren,Zhiguo M,*,Xingwng Zhng,*
a Department of Pharmaceutics,School of Pharmacy,Jinan University,Guangzhou 511443,China
b Department of Chinese Traditional Medicine/Sun-Shengyun Heritage Studio of Eminent TCM Practitioner in Guangdong Province,The First Affiliated Hospital of Jinan University,Guangzhou 510630,China
Keywords:Emodin Liposomes Selenium Stability Sustained release Bioavailability
ABSTRACT Stability of liposomes plays a crucial role in drug delivery,especially in oral aspect.The structural modification of liposomes has been the orientation of efforts to improve their stability and enable the controllability of payload release.This study reported a selenylation strategy to optimize the liposomal structure in an attempt to enhance the nanocarrier’s stability,hence the bioavailability of emodin (EM),an active compound with poor water-solubility.EM-loaded selenized liposomes (EM-Se@LPs) were prepared by thin film dispersion followed by in situ reduction technique.The results showed that EM-Se@LPs were provided with enhancive gastrointestinal stability and exhibited sustained release of drug compared with EM-loaded liposomes (EM-LPs).However,the modified liposomes with Se depositing onto the interior and exterior bilayers did not substantially facilitate absorption of EM.The reinforced structure of liposomes irrelevant to absorption was affirmed to be due to good stability and absorbability of EM itself.Nevertheless,the present work provides an alternative option for stabilization of liposomes instead of conventional methods,which may be promising for oral delivery of physiologically unstable and/or poorly absorbed drugs and systemic drug delivery.
Depending on excellent biocompatibility and biofilm-like property,liposomes have been esteemed as a superior drug delivery system.Liposomes have gained increasing importance in drug delivery,including systemic,oral and local administration [1–3].In terms of all nano-drug delivery systems,liposomes are also the most commercially successful paradigm,resulting in multiple liposomal products approved,for example Doxil®,Amphotec®,and Onivyde®.Although liposomes are qualified with numerous merits,conventional liposomes suffer from some drawbacks as drug delivery carrier,such as poor physiological stability,short retention timein vivo,and premature drug release.Hence,a variety of modification techniques have emerged for liposomes.
The technologies that can be applied for optimizing liposome structure basically involve liposomes coating,solidification and surface modification.Coating the surface of liposomes with functional materials can improve the performances of liposomes both in stability and drug delivery.PEGylation is the most commonly used coating approach [4].Other coating materials available include chitosan,protein,mucin,polyamino acid,polysaccharides,etc.Attachment of coating materials not only changes the structural stability of liposomes,but also modulates the interaction of liposomes with cells and tissues.Solidification refers to the use of solid materials to strengthen liposomes or precipitate on the exterior and interior of liposomes.For instance,calcium alginate andβ-cyclodextrin have been successfully used to stabilize liposomes [5,6].Surface modification deals with the coupling of ligands or antibodies onto the surface of liposomes,which potentially affects their stability andin vivopharmacokinetics.Liposomes anchored with small target molecules also exhibit higher stability,long-circulating time and specific biodistribution [7].Compared with coating and surface modification,the strategy of solidification can ulteriorly improve the stability and achieve sustained/controlled release of liposomes.
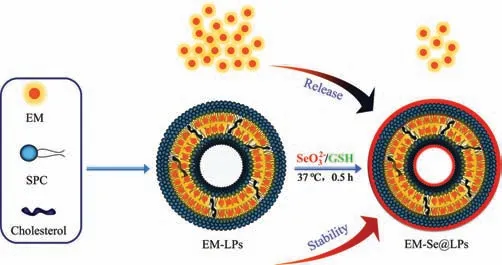
Fig.1.Schematic illustration of selenized liposomes with ameliorative stability and sustained drug release.
Liposome solidification oftentimes shows some technical complexity.Previously,there were only few reports on the gelation of liposomes for stabilization [8,9].It was suggested that gelation could cause the changes in the physicochemical properties of liposomes that improved theirin vitro/in vivoperformance.An alternative approach to liposome solidification is to prepare coreshell polymeric nanoparticles or polymer-lipid hybrid nanoparticles[10,11].With the use of solid polymer,the stability and controllable release of nanocarriers are realized.In theory,metal materials such as aluminum,silver and gold have great advantage in solidifying liposomes.However,these materials were merely used to fabricate non-hybrid nanoparticles,and none of them have been used to solidify liposomes.The toxicity of metal materials is a great challenge for application.By contrast,selenium,a non-metallic micronutrient essential for humans [12],possesses acceptable safety and synergistic therapy with payload,which may be more promising to upgrade liposomes.Herein,we propose the use of selenium to solidify liposomes whereby to improve the stability and release properties of them.
Emodin (EM),a derivative of anthraquinone,is one of the active components in various medical plants such asRheum Palmatum,demonstrating hepatoprotective,anti-inflammatory,antioxidant,antimicrobial and antidiabetic activities [13].However,poor water-solubility (~20 μg/mL) and intestinal adverse reaction build an obstacle to oral administration [14,15].Likewise,selenium exhibits anti-inflammatory,antioxidant and antidiabetic activities,which maybe have therapeutic synergy between them.To this end,it is intended to enhance the oral bioavailability of EM initially through selenized liposomes with their absorption-promoting effect depending on the improved gastrointestinal stability,and then conduct a follow-up study on the synergistic anti-diabetic effect.
In this study,we developed a kind of selenized liposomes(Se@LPs) throughin situreduction technique based on the redox system of glutathione (GSH) and sodium selenite (Na2SeO3).EM-loaded liposomes (EM-LPs) were first prepared by a thin-film hydration method under 25 °C with the solution containing GSH and Na2SeO3(4:1 molar ratio) as hydration medium followed by selenylation at 37 °C for 30 min,resulting in attachment of nascent elemental selenium onto the interior and exterior bilayers of liposomes and generation of EM-loaded selenized liposomes(EM-Se@LPs).Selenylation of liposomes increases its mechanical strength and slows down the payload release as illustrated in Fig.1.
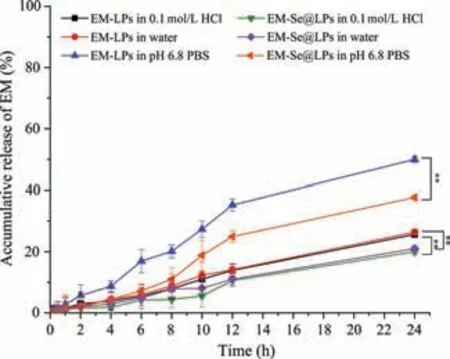
Fig.2.Release profiles of EM from EM-LPs and EM-Se@LPs in 0.1 mol/L HCl solution,pH 6.8 phosphate buffer solution and water.Data expressed as mean ± SD(n=3,**P<0.01,Paired-t-test).
The formulation of EM-Se@LPs was screened with the variables of EM/lecithin ratio,Na2SeO3concentration and reaction time.It was found that the particle size of EM-Se@LPs increased with the increase of EM/lecithin ratio (w/w).Likewise,the particle size of EM-Se@LPs climbed with the increase of Na2SeO3concentration and selenylation time.However,the entrapment efficiency(EE) did not fluctuate distinctly (Fig.S1 in Supporting information).Considering the advantages of small particle size in drug delivery,the preferred formulation of EM-Se@LPs was finalized as 10 mg of EM,400 mg of lecithin,80 mg of cholesterol,10 mL of 0.5 mg/mL Na2SeO3along with quadruple moles of GSH that were formulated in 10 mL of medium and incubated for 0.5 h after hydration.EM-Se@LPs prepared based on the preferred formulation was approximately 126 nm in particle size with a PDI of 0.231,and EM-LPs (counterpart liposomes) possessed a particle size of 109 nm around (PDI 0.247).EM-Se@LPs was slightly larger than non-selenized liposomes (Fig.S2a in Supporting information).It provides evidence that selenium have precipitated onto the interior and exterior phospholipid bilayers of liposomes.Also,theζpotential of EM-Se@LPs (-55.8 mV) slightly differed from that of EM-LPs (-61.2 mV) due to surface deposition of selenium.Besides,EM-Se@LPs and EM-LPs exhibited different appearance and morphology (Fig.S2b in Supporting information).EM-Se@LPs showed a red appearance,whereas EM-LPs were yellow.Both EM-Se@LPs and EM-LPs were spherical in morphology,though EM-Se@LPs exhibited a higher electron-dense corona than EM-LPs,indicating a selenium coverage occurring in liposomes.The physical stability of both liposomes was preliminarily investigated for one week in ambient condition.The particle size did not change significantly with time,but theEEof EM-LPs decreased a little,showing slow drug leakage in conventional liposomes (Fig.S3 in Supporting information).
Ameliorative liposomes stability as a result of selenylation can be perceived from drug release (Fig.2).The release of EM from EM-Se@LPs was significantly slower than EM-LPs whatever in which medium.EM-LPs released approximately 49.97%,25.65% and 26.39% of EM within 24 h in pH 6.8 PBS,0.1 mol/L HCl and water,whereas EM-Se@LPs just released 37.69%,20% and 21%,respectively.The results indicate that Se@LPs can achieve sustainedrelease effect on EM due to the coverage of selenium.This will be favorable for oral delivery of those drugs that are unstable and/or difficult to be absorbed in the gastrointestinal tract,since they can be transported across the absorptive epitheliaviaintact nanoparticles [16,17].
The ability of lipid-based formulation to enhance apparent solubility and oral bioavailability of poorly water-soluble drugs has been broadly confirmed [18–20].Even in the case of lipolysis of lipid carriers,the promoting effect of lipid components on drug absorption is still maintained.There was also evidence that surface modification of lipid carriers could improve its stability and inhibit burst release of drug,thereby enhancing the oral bioavailability of the payload [21].We investigated the oral pharmacokinetics of EM-LPs and EM-Se@LPs and compared with EM suspensions.In surprise,neither EM-LPs nor EM-Se@LPs promoted EM absorption(Fig.3).The relative bioavailability of EM-LPs and EM-Se@LPs to EM suspensions was merely 66.36% and 39.99% respectively (Table S1 in Supporting information),which did not achieve the expected improvement in bioavailability.Generally,drugs that can be promoted for absorption by lipid carriers are mostly highly lipophilic drugs.EM accords with this characteristics,but its intestinal absorption is unknown before.In terms of EM,it may have other gastrointestinal transport behaviors,such as good affinity to enterocytes and no supersaturation after dissolution.This is the first report on lipid formulation failing to promote the oral absorption of poorly water-soluble drug.
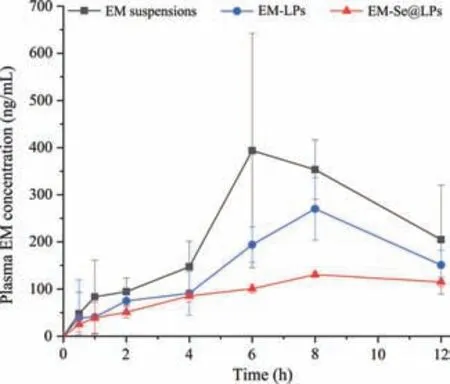
Fig.3.Plasma drug concentration versus time curves of EM suspensions,EM-LPs and EM-Se@LPs.
To explore the underlying absorption mechanisms,we performed the cellular uptake and physiological stability studies on EM,EM-LPs and EM-Se@LPs.Fig.S4 (Supporting information)shows the cellular uptake of free and liposomal EM in Caco-2 cells.It could be found that the cellular uptake rate of free EM was the highest followed by EM-LPs and EM-Se@LPs both at 1 h and 2 h(Fig.S4a).This indicates that EM is well absorbed by the enterocytes itself,which may be associated with its low cytotoxicity(Fig.S5 in Supporting information).At the same concentration,it is easy to understand that free molecules with fine absorbability are more likely to enter cells [22,23].In addition,there was parallel cellular uptake between EM-LPs and EM-Se@LPs,suggesting that selenylation did not significantly change the uptake rate of liposomes.This was corroborated by the parallel cellular internalization between EM-LPs and EM-Se@LPs (Fig.S6 in Supporting information).Nevertheless,EM-LPs and EM-Se@LPs shared different uptake mechanisms.In the presence of transport inhibitors (Fig.S4b),the cellular uptake of EM-LPs and EM-Se@LPs were inhibited to different extent by hypertonic sucrose and chlorpromazine,two clathrin-mediated endocytosis inhibitors.In comparison with EM-Se@LPs,EM-LPs were affected by clathrin-mediated endocytosis more profoundly.Restriction of uptake also occurred under 4°C.This is because cytosis will undergo active deformation of cell membrane that requires expenditure of biological energy.These results manifest that macropinocytosis and clathrin-mediated endocytosis may get involved in the uptake process of EM-LPs and EMSe@LPs [24].
Another factor that affects the oral absorption of lipid carriers as well as their payloads is their gastrointestinal stability [25].The stability study contributes to uncover the mechanism of drug absorptionviathe carrier.The stability of free and liposomal EM in digestive fluids was evaluated using real intestinal juice and simulated gastric/intestinal fluids,including physiological stability,changes in particle size and drug release (Fig.4).As shown in Fig.4a,both free and liposomal EM exhibited good stability in real rat intestinal juice.This is an important reason why EM is well absorbed in the case of suspension formulation,while EM-LPs and EM-Se@LPs cannot promote EM absorption after oral administration.The same thing,marginal drug degradation,happened in the cases of simulated gastric fluids (SGF) and simulated intestinal fluids (SIF) (Fig.4b).These findings prove that EM has no significant intestinal first-pass effect.Lipid carriers including liposomes are readily broken down by digestive enzymes as transport across the harsh gastrointestinal tract [26].In our study,it was found that conventional liposomes (EM-LPs) have stability challenge in the digestive fluids.The particle size of EM-LPs apparently increased upon incubation with SGF and SIF containing gastric lipase and pancreatic lipase,respectively.However,selenized liposomes (EMSe@LPs) showed good resistance against enzymic degradation (Fig.4c).Drug release in SGF and SIF also implied that selenized liposomes have higher stability (Fig.4d).The accumulative release of EM from EM-LPs in SGF and SIG was up to 39.74% and 37.15%within 12 h,respectively,significantly higher than that from EMSe@LPs,which can be attributed to selenylation of liposomes [27].Lipid carriers can promote oral absorption of lipophilic compounds,though they are not a universal platform for any lipophilic drugs.Thein vitroapproaches such as lipolysis model and stability test often fail to adequately predict thein vivoperformance [28].
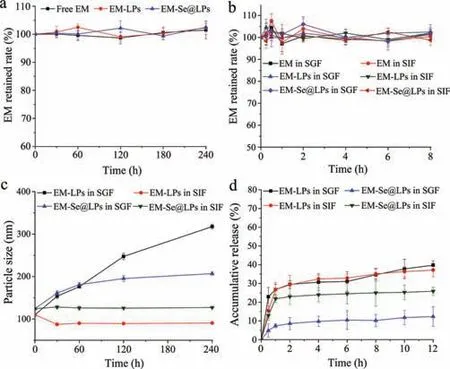
Fig.4.Stability of free and liposomal EM in digestive fluids: (a) In vitro survivability of EM,EM-LPs and EM-Se@LPs in real intestinal juice; (b) in vitro survivability of EM,EM-LPs and EM-Se@LPs in SGF and SIF; (c) changes in particle size of EM-LPs and EM-Se@LPs upon incubation with SGF and SIF; (d) EM release from EM-LPs and EM-Se@LPs in SGF and SIF (n=3,mean ± SD).
In this study,we constructed selenized liposomes with an ameliorative structure for oral delivery of EM aiming to enhance its oral bioavailability.Selenylation does increase the stability of liposomes and achieve sustained drug release.Unfortunately,there was noin vitro-in vivocorrelation between optimized liposomal structure and oral absorption.The underlying reasons lie in good stability and intestinal absorbability of EM itself that eclipse the facilitative effect of liposomes on drug absorption.Although the oral bioavailability of EM has not been enhanced as expected,the present study provides an innovative strategy for solidification of liposomes,which may be suitable for oral delivery of other lipophilic drugs.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This work was supported by the Guangzhou Basic and Applied Basic Research Project (2022).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.04.080.
 Chinese Chemical Letters2023年1期
Chinese Chemical Letters2023年1期
- Chinese Chemical Letters的其它文章
- Diabetic wound healing activated by supramolecular cascade reaction
- MBenes: Two-dimensional transition-metal borides with ordered metal vacancies
- Wet-adhesive materials of oral and maxillofacial region: From design to application
- Diverse catalytic systems for nitrogen-heterocycle formation from O-acyl ketoximes
- Fluorine-containing drugs approved by the FDA in 2021
- The development and application of dual-comb spectroscopy in analytical chemistry