
Regio- and enantioselective conjugate addition of β-nitro α,β-unsaturated carbonyls to construct 3-alkenyl disubstituted oxindoles
2023-03-14 06:52ChangliHeXiaoxueTangXinHeYuqiaoZhouXiaohuaLiuXiaomingFeng
Chinese Chemical Letters 2023年1期
Changli He,Xiaoxue Tang,Xin He,Yuqiao Zhou,Xiaohua Liu,Xiaoming Feng
Key Laboratory of Green Chemistry & Technology,Ministry of Education,College of Chemistry,Sichuan University,Chengdu 610064,China
Keywords:Asymmetric catalysis Conjugate addition Nitroalkenes Regioselectivity Oxindoles
ABSTRACT Reversal of regioselectivity in the catalytic asymmetric conjugate additions of 3-substituted oxindoles to β-nitroenones or β-nitroacrylates was established with chiral scandium catalysts.It enabled the construction of functionalized 3,3-disubstituted oxindoles,including terminal and internal vinyl groups in excellent yields and ee values.
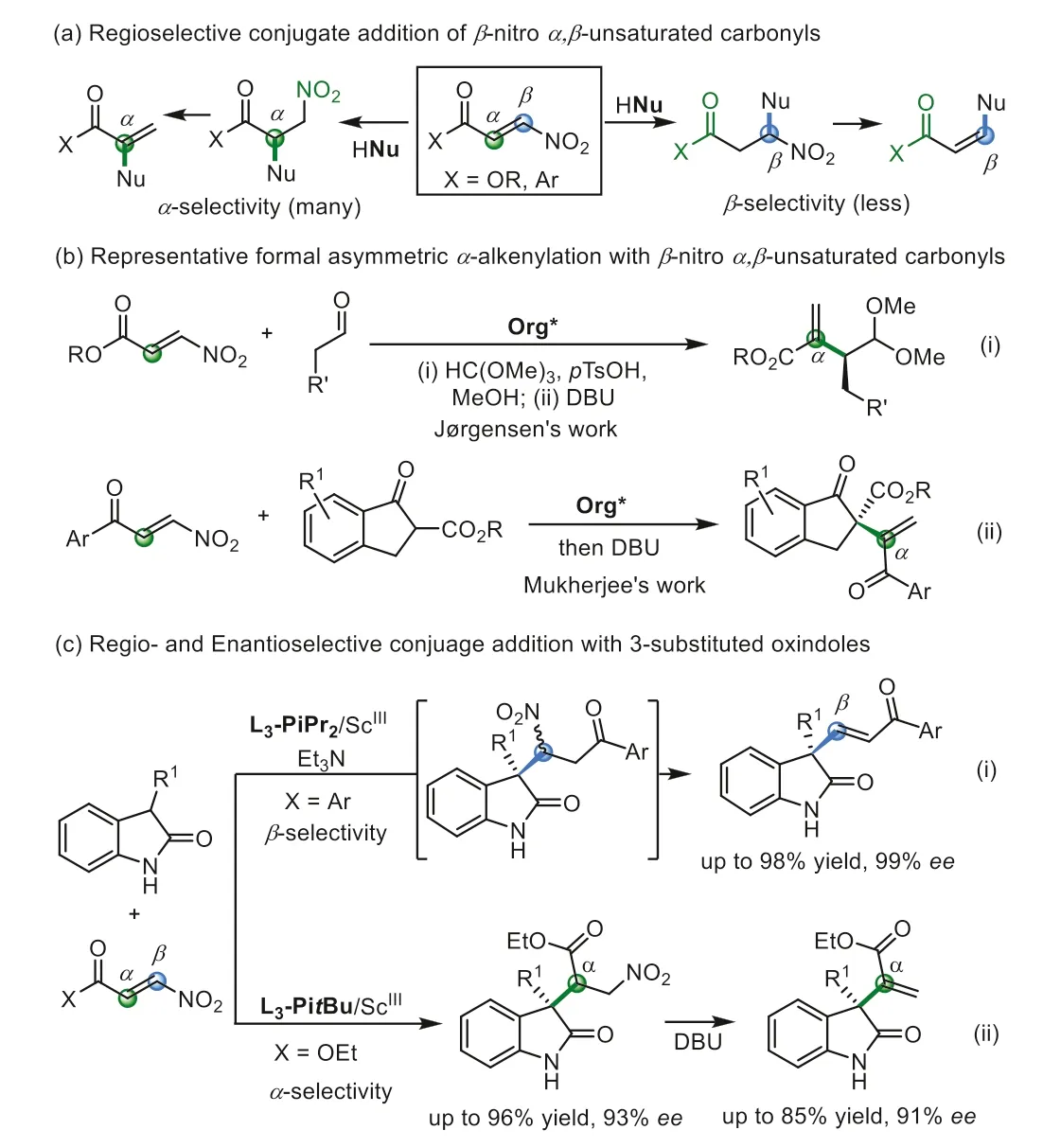
The asymmetric catalytic conjugate addition to electrondeficient alkenes is one of the most useful synthetic routes for the construction of optically active compounds [1–11].Among the various Michael acceptors,the nitroalkenes have drawn more interests not only due to the transformation of nitro group into other functional groups [12–23],but also formation of formal alkenylation derivativeviathe elimination under suitable reaction conditions [24–27].In recent years,the asymmetric catalytic conjugate of rich functionalizedβ-nitroα,β-unsaturated carbonyls have been studied (Scheme 1a),which has higher reactivity as pullpull olefins due to the global electron depletion [28,29].Nevertheless,the doubly activated olefins with different vicinal electronwithdrawing groups might raise the regioselectivity.Hu’s group reported chiral RhI-diene catalyzed asymmetric three-component reaction of aryldiazoacetates,aromatic amines andβ-nitroacrylates to obtainγ-nitro-α-amino succinates [30].Asymmetric conjugate addition of metal alkyl reagents or arylboronic acids toβnitroacrylates to generateβ-amino acid precursors was also realized by several research groups [31–34].On the other hand,formalα-alkenylations of aldehydes orβ-ketoesters have been reported by Jørgensen [35] and Mukherjee [36],respectively,usingβ-nitroacrylates (Scheme 1b,i) andβ-nitroenones (Scheme 1b,ii).However,in those cases,α-selective conjugate addition occurred which might provide a bias that this is the dominated regioselectivity in nucleophilic addition of pull-pull olefines.
Oxindoles represent a privileged backbone existing in many natural products and bioactive compounds,and also important building blocks in organic synthesis [37–43].The nucleophilic conjugate addition of 3-substituted oxindoles increased the diversity of oxindoles,and the use of but–2-ynedioate [44,45],alkynones [46],orβ-haoloalkenes [47] as the Michael acceptors could give vinylic substituted oxindoles.Nevertheless,asymmetric alkenylation to construct quaternary oxindoles with anexo-methylene-substitution is elusive.In view of the biological properties of oxindoles,the interesting regio-selectivity of nitroolefins,as well as the elimination tendency to generate formal alkenylated product,we carried out regio-and enantioselective conjugate addition ofβ-nitroα,β-unsaturated carbonyls with 3-substituted oxindoles.Here,we found that in the presence of chiralN,N′-dioxide/scandium complexes [48–52],the reaction withβnitroenones occurredviaaβ-selective addition,in which benzoyl group acts as a stronger directing group (Scheme 1c,i); whereas the reaction withβ-nitroacrylates occurredviaaα-selective addition where nitro-group acts as the activated group (Scheme 1c,ii).Both terminal and internal vinyl substituted oxindoles could be obtained in excellent yield and enantioselectivity in the presence of suitable bases.
In the initial study,we chose phenylβ-nitroenone B1 as the Michael acceptor and 3-benzylindolinone A1 as the nucleophile to construct quaternary carbon center (Table 1).Firstly,metal salts coordinating with chiralN,N′-dioxide ligand L3-PiPr2(Fig.1) which was derived from L-pipecolic acid,were investigated in the presence of Na3PO4as a base in CHCl3at 30 °C (see Supporting information for details).It was found that Mg(OTf)2,Ni(OTf)2,and Yb(OTf)3could promote the reaction while Sc(OTf)3was more effective to yield the formal alkenylation product (E)-C1via βselective conjugate addition/nitro-elimination in 81% yield and 94%ee(entries 1–4),with different regio-selectivity from Mukherjee’s report (Scheme 1b,ii).For ligand skeletons (see Supporting information for details),it seemed that both the amino acid backbone and the steric hindrance of the amide subunits had dramatically influence on the reactivity and enantioselectivity.The ligand of L3-PrPr2(L-proline based one) and L3-RaPr2(L-ramipril based one)gave inferior results than L3-PiPr2(entries 4–6).Decreasing the steric hindrance of the 2,6-disubstituted amide fromiPr to Et or Me group led to dropped reactivity and enantioselectivity (entry 4vs.entries 7 and 8).Bases affected the reactivity a little with slight change of the enantioselectivity (entries 9–11; see Supporting information for details).When the reaction was carried out with Et3N instead of Na3PO4,97% yield with 98%eewas obtained after 4 h (entry 11).At 5 mol% of catalyst loading,the enantioselectivity of the reaction could be maintained but the yield dropped a little (entry 12).If without a base,only 16% yield was obtained(entry 13).
Scheme 1.Reaction about β-nitroenones and β-nitroacrylates.
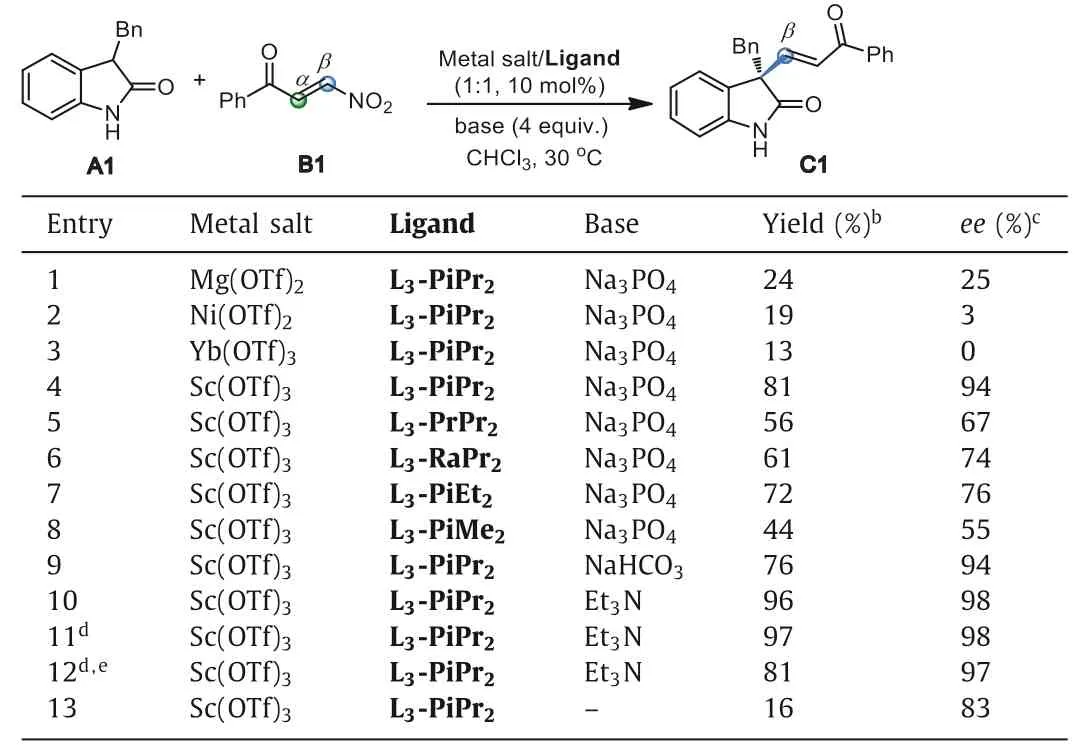
Table 1 Optimizations of reaction conditions with β-nitroenone.a
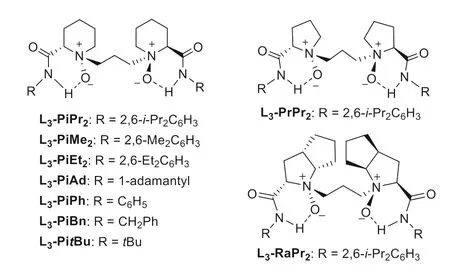
Fig.1.Chiral N,N′-dioxides used.
With the optimized reaction conditions in hand (Table 1,entry 11),the substrate scope was evaluated (Scheme 2).Substituents atpara-position of the benzyl group had little effect on the reactivity and enantioselectivity,regardless of the electron-donating groups or electron-withdrawing groups (70%-98% yields with 90%-98%ee,C2-C8) except for 4-NO2-Bn (C4; 70% yield with 90%ee).Substituents atortho-position ormeta-position gave almost the same excellent enantioselectivities (80%–96% yields,95%-99%ee,C9-C16 and C23),but the 2,6-dichlorobenzyl-substituted C17 was isolated in slightly lower yield (63%) with 94%ee.In particular,3-substituted oxindoles,including methyl,2-naphthylmethyl,2-thienylmethyl,or 3-thienylmethyl,all were efficient nucleophiles to deliver the corresponding quaternary oxindole derivatives (C18,C20-C22) in 65%-87% yields with 86%-97%ee.In comparison,3-phenyl substituted one afforded lower yield andeevalue (C19,30%yield and 74%ee).The absolute configuration of the product C9(CCDC: 2117166) was determined to be (R,E) by single-crystal X-ray diffraction analysis (for details,see Supporting information).
Different arylβ-nitroenones with diverse electronic and steric nature could be tolerated to generate the desired alkenylated products C24-C29 in high yields with excellenteevalues (84%-93%yields,96%-98%ee).β-Nitroenones bearing 2-naphthyl,1-naphthyl or 2-thienyl group,gave the products C30-C32 in moderate to high yields with good enantioselectivities (65%-98% yields,94%-98%ee).A scale up synthesis of internal vinyl substituted oxindole C1 was accessible without obvious influence on the reactivity and enantioselectivity (92% yield and 97%ee).
To investigate the influence of pull-substitution on the regioselectivity of dissymmetrical pull-pull alkenes,we tested the reaction between 3-substituted oxindole A1 andβ-nitroacrylate D1.However,as shown in Table 2,under the above optimal condition using L3-PiPr2/Sc(OTf)3and Et3N,α-selective addition product E1 and its diastereoisomer E1′were observed in good yield but with dramatically dropped enantioselectivity (Table 2,entry 1).The use of otherN,N′-dioxide ligands (see Supporting information for details),such as L3-PrPr2and L3-RaPr2,gave nearly equal amount of diastereoisomers in poor enantioselectivity (entries 2 and 3).Interestingly,when less steric aniline-based L3-PiPh,or phenylmethanamine-based L3-PiBn used (Fig.1),better enantioselectivity for every product was observed (entries 4 and 5).When aliphatic amine-based ligands employed,the yields of major product E1 increased obviously (entries 6 and 7).Especially,the tertbutyl amine-based ligand L3-PitBu gave the product E1 in 58% yield with 87%ee,and the product E1′in 23% yield with 82%ee(entry 7).Less amount of base is beneficial to the formation ofα-selective product E1 (entries 8 and 9),which could be isolated in 75% yield with 93%eewith 0.1 equiv.of Et3N after prolonged reaction time at 0 °C (see Supporting information for details),and the minor diastereoisomer E1′was also obtained in 21%yield with 92%ee(entry 9).While if without base,the reaction did not occur and in the absence of DBU the nitro-group maintained in the products without elimination.The formal alkenylation productsviaelimination of nitro-group in the presence of DBU could be afforded without obvious erosion of the enantioselectivity from bothα-selective addition product E1 and E1′(Eq.1),yielding the same formal alkenylation product F1 with identical absolute configuration which showed that the diastereoselectivity raised from theα-position of nitroacrylate.
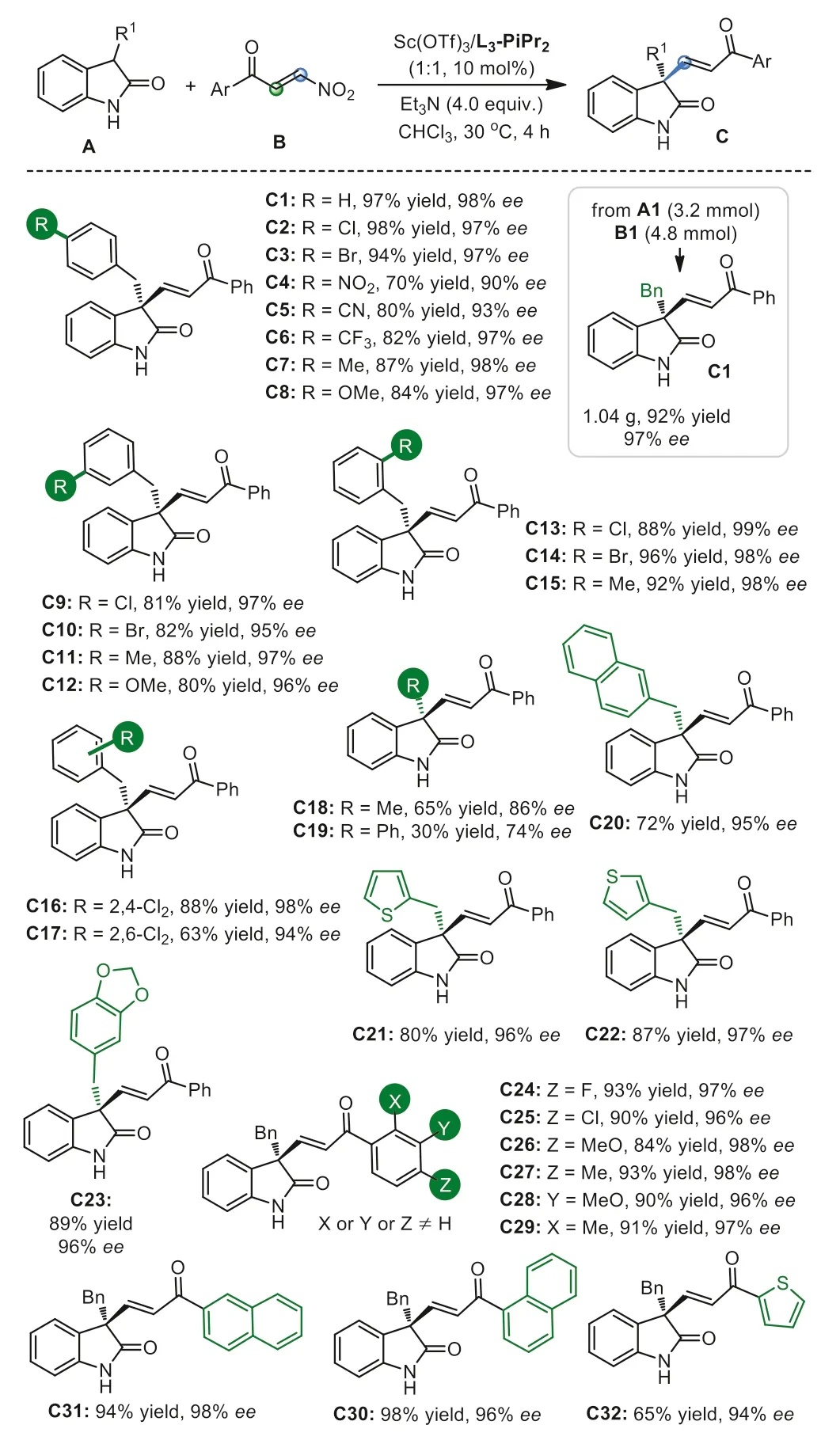
Scheme 2.Substrate scopes of the reaction with β-selective addition.Unless otherwise noted,all reactions were carried out with Sc(OTf)3/L3-PiPr2 (1:1,10 mol%),Et3N (4.0 equiv.),A (0.10 mmol) and B (0.15 mmol) in CHCl3 (1.0 mL) at 30 °C for 4 h.Yield was determined by Isolated yields. ee was determined by HPLC on a chiral stationary phase.
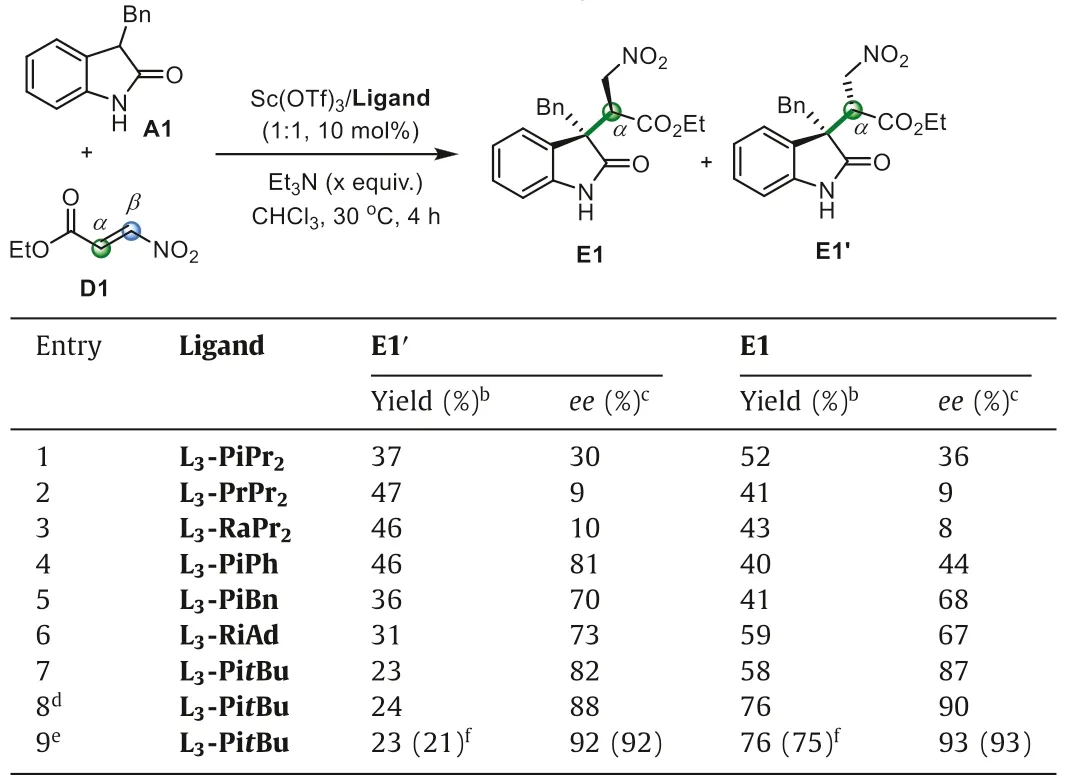
Table 2 Optimization of the reaction conditions with β-nitroacrylate.a
Having the optimal reaction conditions in hand (Table 2,entry 9),the scope of N–H 3-substituted oxindoles was evaluated(Scheme 3).Both electron-rich and electron-poor substituents at the phenyl group of 3-benzyl oxindoles were tolerated,furnishing theα-selective conjugate addition products E2-E9 as the major products (63%-76% yield with 84%-92%ee),and the adducts E2′-E9′as the minor (15%-22% yield,81%-92%ee).The electronrich substituents gave slightly lower yield ofα-selective adducts compared with the electron-withdrawing ones.2-Naphthylmethyl oxindole was also tolerable,even though a moderate yield was obtained (58% yield with 86%eefor E10,and 17% yield with 90%eefor E10′).For 3-thienylmethyl oxindole,the corresponding product E11 was isolated in 70% yield with 88%ee.When DBU was added to the finished asymmetric catalytic reaction mixture,the nitro eliminationexo-methylene oxindoles F1-F11 could be obtained in 73%-85% yields with 86%-91%ee.A gram-scale synthesis ofexomethylene oxindole F5 in 80% yield and 88%eedemonstrated the reliability of this method.
We performed calculation about the natural bond orbital charges computed at the M06–2X/Def2-SVP level to unravel the factors controlling the regioselectivity ofβ-nitroenone B1 andβnitroacrylate D2 [53].As shown in Fig.2a,theβ-carbons of both B1 and D2 have stronger positive charges which seem to be more likely to undergo nucleophilic attack,while the latter is inconsistent with the experimental results.It implies that it is difficult to evaluate the regioselectivity of dissymmetrical pull-pull alkenes in nucleophilic addition simply relying on natural bond polarizations,and the regioselectivity relies on entangled factors involving the interaction with the catalyst,steric hindrance,stability of the intermediates,and others.We rationalized that the reversal of regioselectivity in the addition reaction ofβ-nitroenone B1 withβ-nitroacrylate D1 might be due to the selective coordination of the three kinds of pull groups,following the precedence as benzoyl>nitro>ester with the scandium catalyst.The Lewis acid activations occur at the benzoyl group ofβ-nitroenone B1,while at the nitro group ofβ-nitroacrylate D1,respectively,lowing the LUMO energy of the Michael acceptors to benefit theβ-selective conjugate addition andα-selective conjugate addition,respectively.
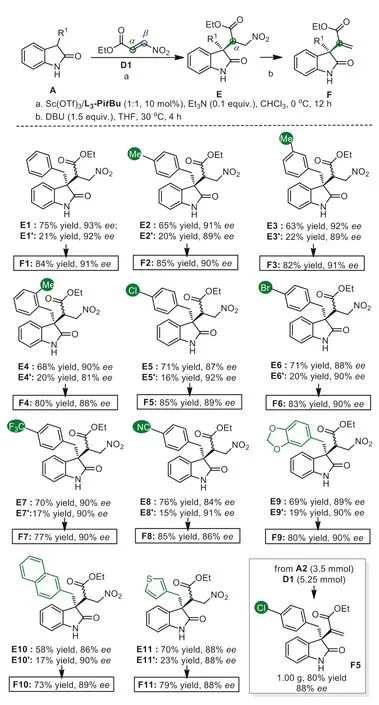
Scheme 3.Substsrates scopes of regio- and enantio-selective reactions of βnitroacrylate.Unless otherwise noted,all reactions were carried out with Sc(OTf)3/L3-PitBu (1:1.1,10 mol%),Et3N (0.1 equiv.),A (0.10 mmol) and D1(0.15 mmol) in CHCl3 (1.5 mL) at 0 °C in air for 12 h.For the synthesis of formal alkenylation product F,DBU (1.5 equiv.) was added after the conjugate addition process finished.Yield was determined by isolated yields. ee was determined by HPLC or SFC on a chiral stationary phase.
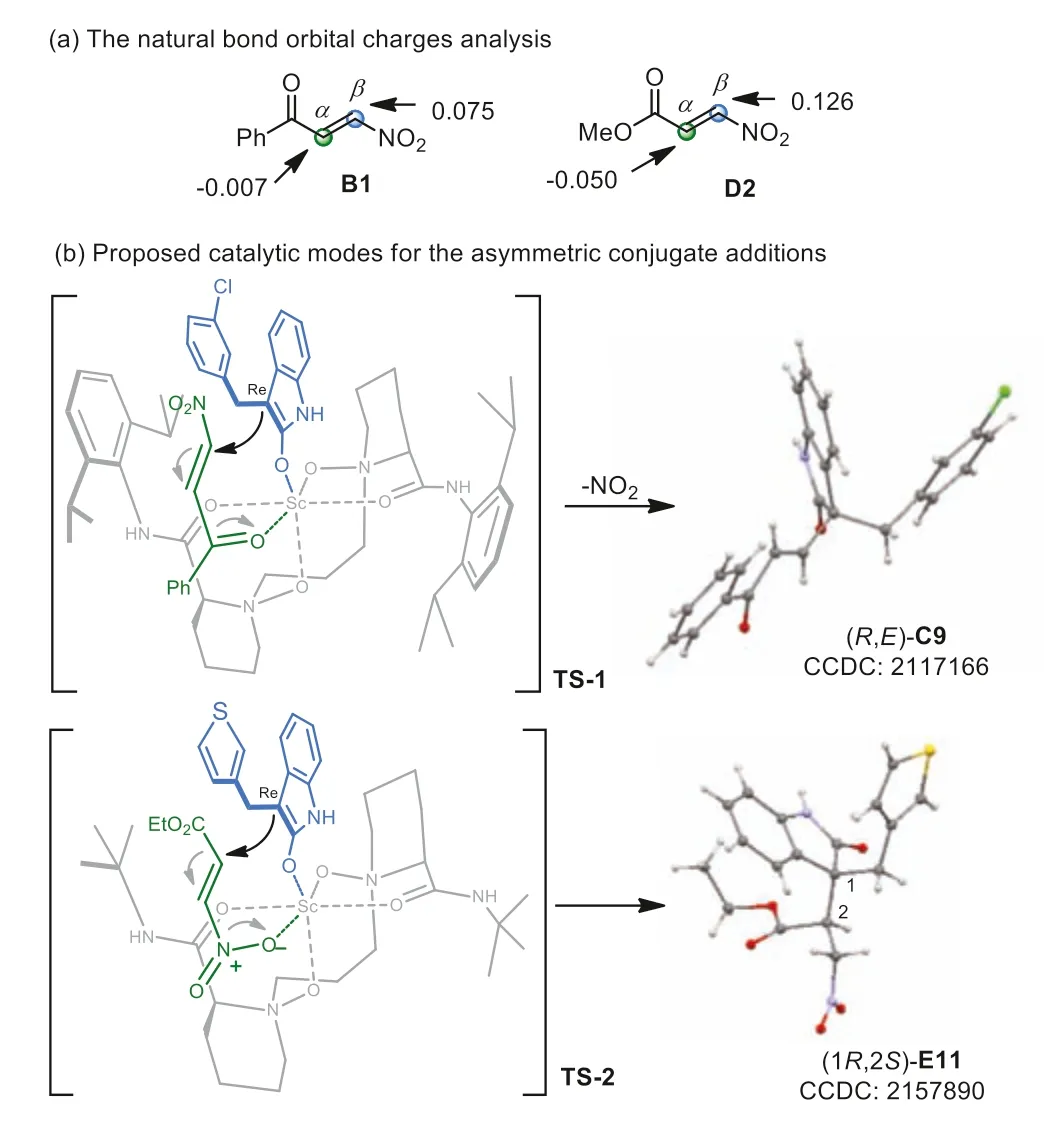
Fig.2.Mechanism consideration.
The absolute configurations of the two products were determinedviaX-ray crystal analysis (for details,see Supporting information,pages 12–16),which showed the same stereoselectivity at the quaternary carbon center,indicating the same facial selectivity for the nucleophilic addition of the 3-substituted oxindoles.Based on the general catalytic behaviors of chiral Lewis acids ofN,N′-dioxide ligands [48–52],we proposed a plausible possible catalytic modes to rationalize the regio- and enantioselectivity of the reaction.The four oxygens ofN,N′-dioxide bond to the scandium center to form the chiral Lewis acid catalyst.As shown in the transition state TS-1 of Fig.2b,the benzoyl group ofβ-nitroenone B1 coordinates to the metal center at the front site and the enolized oxindole bonds at the top site,leaving itsRe-face toward theβnitroenone.TheRe-face of the Michael acceptor is blocked by the left amide subunit of the ligand,undergoing the addition with itsSi-face to yield theβ-selective addition intermediate.Upon elimination of the nitro group the formal alkenylation product (R,E)-C9 was obtained as the major product.Similarly,whenβ-nitroacrylate D1 is used as the Michael acceptor,the nitro group prefers to coordinate to the scandium center over ester group,and as shown in TS-2 the enolized oxindole follows the similar coordination to the above TS-1.Thus,theα-selective adduct (1R,2S)-E11 is generated as the major isomer.The supplementary crystallographic data of E11 (CCDC: 2157890) can be obtained free of charge from The Cambridge Crystallographic Data centre.
To sum up,a highly efficient asymmetric Michael reaction ofβ-nitroα,β-unsaturated carbonyls with N–H 3-substituted oxindoles was realized by using chiralN,N′-dioxide/Sc(OTf)3complexes.The installation of internal or terminal vinyl substitution at the C3-position of oxindole was accessiblevia β-selective conjugate addition ofβ-nitroenones,andα-selective conjugate addition ofβnitroacrylates,respectively.The selective coordination of the scandium catalyst with pull-group of nitroolefins might account for the reversal of regioselectivity.Our protocols provided a useful route for the construction of functionalized quaternary oxindoles.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
We appreciate financial support from the National Natural Science Foundation of China (No.21890723).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.05.001.
 Chinese Chemical Letters2023年1期
Chinese Chemical Letters2023年1期
- Chinese Chemical Letters的其它文章
- Diabetic wound healing activated by supramolecular cascade reaction
- MBenes: Two-dimensional transition-metal borides with ordered metal vacancies
- Wet-adhesive materials of oral and maxillofacial region: From design to application
- Diverse catalytic systems for nitrogen-heterocycle formation from O-acyl ketoximes
- Fluorine-containing drugs approved by the FDA in 2021
- The development and application of dual-comb spectroscopy in analytical chemistry