
牙龈卟啉单胞菌与常见非肿瘤性疾病的研究进展*
2023-03-10 01:58买热拍提买明热孜万姑丽亚森综述郭治辰龚忠诚审校
重庆医学 2023年4期
买热拍提·买明,热孜万姑丽·亚森 综述,郭治辰,龚忠诚△ 审校
(1.新疆医科大学第一附属医院口腔颌面肿瘤外科,乌鲁木齐 830054;2.新疆维吾尔自治区口腔医学研究所,乌鲁木齐 830054;3.西安交通大学口腔医院,西安 710004)
牙龈卟啉单胞菌(P.gingivalis)口外感染方面的相关研究最早可追溯至19世纪80年代,其证实了P.gingivalis涉及多种系统性疾病,如心血管疾病、神经系统疾病、自身免疫性疾病、呼吸系统疾病、代谢性疾病、不良妊娠等。现将P.gingivalis与常见非肿瘤性疾病的研究进展综述如下。
1 P.gingivalis的主要特征
P.gingivalis是一种革兰氏阴性专性厌氧菌,最初作为噬纤维菌-黄杆菌-拟杆菌(CFB)群的模式生物引起了学者兴趣,后来作为口腔病原体渐渐露出水面,随着P.gingivalis参与阿尔茨海默病(AD)的证据被发现,临床对其关注度达到高峰。慢性牙周炎(PD)是指牙周围组织因细菌积聚(牙菌斑形成)而发生的炎症,可使牙齿与牙槽骨的胶原附着逐渐丧失,如不加以控制,可能导致牙齿松动、脱落[1]。P.gingivalis是公认的口腔疾病牙周炎主要病原体,其致病因素多样,包括自身结构成分[如脂多糖(LPS)]、菌毛、热休克蛋白(HSP)和分泌成分(如牙龈素和外膜囊泡)[2]。这些因子刺激相邻上皮细胞产生各种细胞因子,包括白细胞介素(IL)和肿瘤坏死因子-α(TNF-α)[3]。P.gingivalis同时是一种兼性胞内细菌,能入侵多种宿主细胞,包括牙龈上皮细胞、内皮细胞、血管平滑肌细胞(VSMCs)、树突状细胞(DC)和神经元[4]。有关P.gingivalis口外感染方面的研究最早源于19世纪80年代,已证实P.gingivalis涉及多种系统性疾病。P.gingivalis产生的牙龈素会导致局部组织破坏,扰乱宿主的抗菌防御,导致口腔和肠道内共生细菌的过度生长及播散。这会使有毒代谢物过度产生,促进心血管疾病和自身免疫相关代谢紊乱。大脑中产生的牙龈素则对AD的发生、发展有重要意义。P.gingivalis产生的肽基精氨酸脱亚胺酶(PAD)亦是其重要致病因素[5],其可打破瓜氨酸蛋白的免疫耐受,加重易患类风湿性关节炎(RA)个体的症状[5]。
囊泡是细胞膜的一部分,大小为50~300 nm,形成于A-LPS和具有C-末端结构域(CTD)的蛋白质[如牙龈素、PAD或血红素结合蛋白35(HBP35)]组成的双层膜结构[2]。菌毛促使细菌黏附宿主细胞、与外基质分子、细胞因子和纤维蛋白原、CD14、β2整合素、toll样受体(TLR)2、TLR4结合[6-7]。这些相互作用促使单核细胞趋化蛋白-1(MCP-1)、TNF-α、IL-1β、IL-6、IL-8等促炎因子合成,并诱导多细胞间黏附分子-1(ICAM-1)、血管细胞黏附分子-1(VCAM-1)[6]、P-选择素、E-选择素、CD40、CD80和CD86的表达[6-7]。其中,IL-8能够诱导可结合血红蛋白的受体(HbR),以捕获卟啉环和血红素,从而得到P.gingivalis生长所需的铁[8]。LPS结合高度敏感的模式识别受体(PRR)后通过核因子-κB(NF-κB)依赖性产生TNF-α,激发涉及胞外调节蛋白激酶(ERK)的炎症,激活p38和c-Jun氨基末端激酶(JNK),上调纤溶酶原激活物抑制剂(PAI)-1的表达[9],因此,LPS对细胞串扰作用、P.gingivalis的毒力至关重要。P.gingivalis与牙周来源致病菌能够在到达血液之前迁移并侵入上皮、结缔组织[4],其内毒素和外毒素也能到达血液,从而扩散到远处发挥毒性。上述某些促炎分子,如IL-1β、IL-6或TNF-α会进入体循环,触发肝脏等其他组织的反应[10]。
2 P.gingivalis与动脉粥样硬化(AS)
AS是一种常见于大中型动脉的慢性疾病,被认为是大多数心血管疾病的发病基础[10-12]。在过去数十年中,相关证据越来越倾向于AS是一种慢性炎症疾病。动脉斑块形成是AS的病理基础,其由脂质、钙、巨噬细胞等成分在动脉壁堆积而成。患者因外科手术、牙龈炎、口腔溃疡、口腔黏膜病变和创伤等使血管破裂,引起口腔中的病原体随血液循环迁移,特别是P.gingivalis[10]。2012年,美国心脏协会发表了声明[11],支持PD与动脉粥样硬化性血管疾病(ASVD)之间的联系,该联系独立于已知的混杂因素。
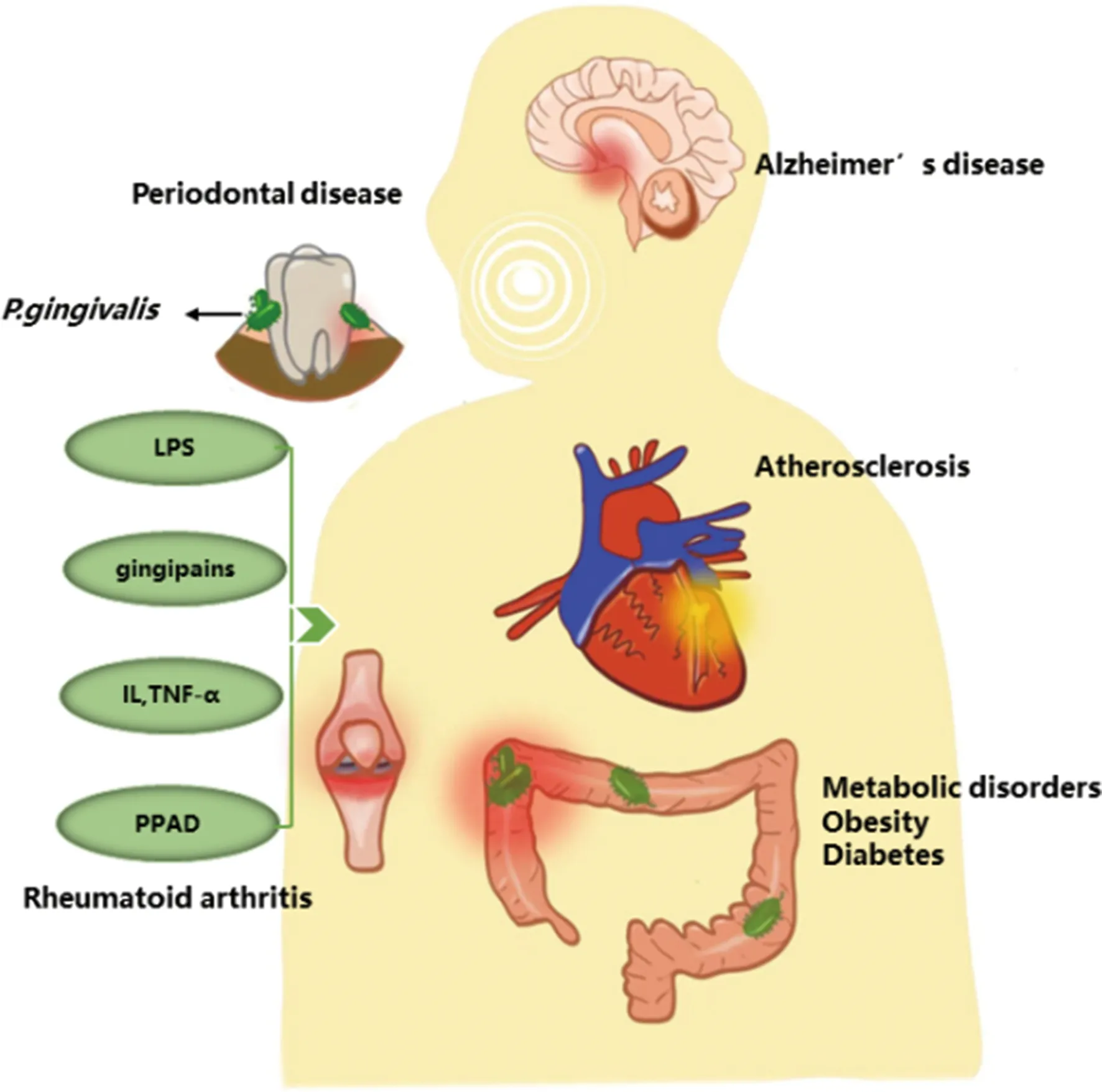
图1 P.gingivalis与全身多种疾病的相关性
P.gingivalis可通过连接、进入/内化、传输、持久化和退出等过程入侵心血管细胞和组织[13],其在AS中的作用机制可归纳为以下多个方面。(1)P.gingivalis可导致内皮细胞功能障碍。内皮氧化应激促进单核细胞黏附和促炎细胞因子释放,导致内皮功能障碍,这是AS的前兆[14]。P.gingivalis能够诱导严重的内皮细胞氧化应激[15],这一过程主要由TLR-NF-κB信号轴促进。P.gingivalis表面LPS被TLR介导识别后,激活下游信号通路NF-κB及其活性亚基p65,从而触发氧化应激[9,16]。氧化应激后,P.gingivalis会触发脉管系统的炎症反应,内皮细胞中的促炎因子IL-1β、IL-6、TNF-α和γ-干扰素(IFN-γ)分泌增加[17],同时IL-1β、IL-8和MCP-1等细胞因子促进人主动脉内皮细胞(HAEC)中产生趋化因子[18]。P.gingivalis上调的IL-8可通过激活NF-κB通路直接增加内皮通透性[19];IL-1β通过NOD样受体热蛋白结构域相关蛋白3(NLRP3)炎症小体激活释放,并促进动脉粥样硬化斑块的破裂。NLRP3为核苷酸结合寡聚化结构域样受体家族含pyrin域3的炎症小体,在AS的病理生理学中起重要作用,P.gingivalis可通过炎性环境中的巨噬细胞激活NLRP3炎性小体,推进内皮中的炎症过程和氧化应激[20]。P.gingivalis还能够下调生物种基因BMAL1的表达,促进氧化应激,加重动脉粥样硬化[16]。研究报道,不同P.gingivalis菌株引起不同程度的内皮功能障碍[13,18],如P.gingivalis381(菌毛Ⅰ型)诱导菌毛依赖性现象,如人冠状动脉内皮细胞中GroA、GroE、IL-6、IL-8基因的表达、TLR介导VCAM-1和内皮白细胞黏附分子-1(ELAM-1)的分离。W83有包膜但没有菌毛,可适度激活TLR2并减弱炎症反应[13]。此外,P.gingivalis能够诱导内皮细胞凋亡和内皮-间充质转化(EndMT),并抑制其增殖,从而减少内皮细胞的数量,导致血管内皮屏障的损伤[21]。此时,循环中的白细胞和低密度脂蛋白(LDL)在受损的内皮下积聚,导致AS的发生[22],此过程与巨噬细胞通过CD36摄取和积累脂蛋白相关[20]。(2)P.gingivalis及其毒力成分(如LPS、菌毛)参与AS形成过程中单核细胞活动的各个阶段,支持单核细胞向内皮表面迁移、内膜浸润,并分化为M1型巨噬细胞,最终形成泡沫细胞,而泡沫细胞对AS的发展至关重要[21]。P.gingivalis通过巨噬细胞的内化实现免疫逃避,继而保持毒力和持续性感染[21]。巨噬细胞对P.gingivalis的摄取取决于3型补体受体[CR3(CD11b/CD18)]和TLR2[23]。(3)VSMCs对动脉损伤和脂质浸润的反应。LPS可刺激VSMCs增殖和钙化,导致血管钙化[24-25]。牙龈素对血浆的胰蛋白酶样作用有助于VSMCs增殖,这一过程可能主要由转化生长因子-β(TGF-β)/Notch途径调节[25]。(4)P.gingivalis影响粥样斑块中的免疫细胞。P.gingivalis及其LPS和牙龈素可以激活单核细胞,促进Th17/IL-17反应,使TNF-α、IL-1β、IL-6和IL-17增加,经TLR2/TLR4信号介导,促进炎症反应,诱导动脉粥样硬化斑块形成[26]。
P.gingivalis感染损伤内皮细胞,导致泡沫细胞的形成及平滑肌细胞的钙化,随Th/Tregs失衡、内皮细胞活化、斑块形成,对AS的进展起促进作用。
3 P.gingivalis与类风湿
研究表明[27],PD患者RA患病率远高于非PD患者,且关节炎和牙周炎的严重程度呈正相关。RA的病因非常复杂,大多数病例是由瓜氨酸化蛋白的自身免疫反应引发,蛋白质的瓜氨酸化通过PAD进行。虽是生理条件下产生,因遗传易感体的免疫耐受性丧失,导致滑膜中产生抗瓜氨酸合成蛋白的自身抗体(ACPA),由此发展为RA[20,28]。P.gingivalis能够打破机体对瓜氨酸化蛋白的免疫耐受[29]。
此外,P.gingivalis使肠道生态失调,上调IL-17水平,从而促进RA的进展[5]。Th17/Treg的失调也起重要作用,P.gingivalis通过诱导TLR2和IL-1驱动的Th17细胞反应激活免疫系统[29]。在抗原诱导的C57BL/6小鼠关节炎模型中,Th17/IL-17A信号通路参与了P.gingivalis诱导的实验性关节炎,导致关节中性粒细胞浸润增加[30]。P.gingivalis感染的抑制胶原性关节炎模型(CIA)小鼠中,产生IL-10的调节性B细胞(B10)被下调,P.gingivalis直接促进Th17、Treg和骨髓源性抑制细胞(MDSCs)的分化,但抑制B细胞向B10细胞分化,提示B10细胞的下调可能是PD促进RA的关键机制,其他主要免疫抑制细胞如Treg和MDSCs等均被下调[28]。同时,P.gingivalis感染促进破骨细胞分化,加入补体C5a中和抗体则抑制其分化;与P.gingivalis抗体阴性的RA患者比较,阳性的RA患者血清C5a水平升高,表明P.gingivalis感染通过C5a促进RA的进展[31]。PAD诱导蛋白质的瓜氨酸化,一直处于RA和PD相关致病假说的中心。P.gingivalis毒力因子(如LPS、菌毛等)可被宿主口腔内的上皮细胞、DC、中性粒细胞的TLR、蛋白酶激活受体(PARS)和/或NOD2受体识别,释放炎性细胞因子和趋化因子,激活补体系统、RANKL信号通路和T细胞的分化[32],最终促进破骨细胞的产生。RA的慢性骨侵蚀和PD的慢性牙槽骨破坏的炎症细胞和促炎细胞因子相似,PD可能是RA中发生的自身免疫炎症反应启动和维持的一个因素[33]。
4 P.gingivalis与AD
AD是一种神经退行性疾病,其特征是进行性认知下降和记忆丧失,最终导致智力完全丧失甚至死亡[33]。AD的病理学主要有两个特征:由神经原纤维缠结组成的高磷酸化tau蛋白沉积和淀粉样蛋白β(Aβ)沉积[34]。一项队列研究发现,PD患者患AD的风险升高[34]。动物实验表明,静脉感染P.gingivalis在SD大鼠中诱导tau过度磷酸化[35]。此外,连续5周系统地暴露于P.gingivalis-LPS会导致12月龄野生型小鼠脑中Aβ蛋白的聚集,出现学习和记忆障碍,此过程可能通过TLR4/NF-κB信号通路来实现。糖原合酶激酶3β(GSK3β)作为tau蛋白的激酶[36],参与P.gingivalis-LPS促进的神经炎症和tau过度磷酸化[36-37],而P.gingivalis产生的牙龈素协同促进小胶质细胞向感染部位迁移,激活PI3K/Akt和MEK/ERK通路,从而加速AD进展[38]。P.gingivalis在局部诱导产生的促炎介质如TNF-α、IL-6和IL-1β可通过血脑屏障或通过调节血脑屏障通透性到达中枢神经系统[25,33]。此外,研究证实P.gingivalis会影响Aβ蛋白的清除,并诱导免疫抑制[38]。P.gingivalis会进入细胞质,逃避宿主的免疫监视并持续释放炎性介质。其毒力因素抑制宿主整体的免疫反应,使宿主细胞释放大量促炎细胞因子,导致机体长期处于炎症状态,影响tau蛋白的合成与代谢,使Aβ淀粉样蛋白在脑组织沉积,引起认知障碍和记忆力减退。
5 P.gingivalis与不良妊娠结局
不良妊娠结局是一个宽泛术语,主要包括先兆子痫(妊娠20周后伴有蛋白尿或肺水肿、少尿或惊厥的妊娠高血压)、宫内感染(由生殖道或非生殖道来源的微生物引起)、早产(妊娠第37周前的任何活产)、低出生体重(新生儿体重<2 500 g)、自然流产(妊娠20周前流产)和/或死产(新生儿出生时无生命迹象)等[39]。有学者于1996年报道,母亲患有牙周炎可能是不良妊娠结局的潜在危险因素[40]。
相对于牙周组织健康的孕妇,患有牙周炎的孕妇的胎盘可能携带各种牙周病原体[41]。P.gingivalis的LPS在合胞滋养层、滋养层细胞、蜕膜细胞、脐带、羊膜上皮和羊水、胎盘内皮细胞、胎儿毛细血管和新生儿鼻胃液中均可被检测到[42]。临床研究表明,在不良妊娠的女性中,龈沟液中的炎症细胞因子水平升高,这可能会加速分娩[43]。动物模型研究提示,口腔内持续感染P.gingivalis后,母体血清中TNF-α、IFN-γ增加2.5倍,IL-17、IL-6、IL-1β增加2倍,且胎盘组织中TLR2和Fas/Fas配体途径被上调,导致早产和低体重出生儿[39]。研究表明,P.gingivalis通过JNK和p53/p38通路,导致人滋养层细胞损伤,这是扰乱细胞周期和促进凋亡的重要因素,会对正常妊娠产生负面影响[42]。此外,LPS-TLR4相互作用增加了子宫肌层中Rho相关蛋白激酶的活性,使子宫肌细胞中钙的内流,从而促进肌层收缩,增加了复发性流产的风险[42]。P.gingivalis上调了与白细胞上补体受体结合的C5a水平,并损伤白细胞杀伤能力[31]。P.gingivalis可持续感染和长期存活在胎儿和母体组织中,逃避机体的免疫监视,引起微生物菌群失调;其独特的毒力因子、表面黏附能力、产生的相关酶能直接损伤胎儿和母体组织的形态;其所产生的促炎细胞因子、自由基和相关酶蛋白增加,Th17/Tregs失衡,从而增加子宫肌层收缩和早产风险。此外,P.gingivalis增加胎儿肾上腺皮质醇的生成[42]。在此基础上,必要时可行进一步研究,评估P.gingivalis感染是否是妊娠和胎儿并发症的绝对/相对危险因素。
6 P.gingivalis与肥胖症
口腔和肠道微生物群中的厌氧菌在调节宿主代谢平衡和内环境稳定中发挥了重要作用,也通过宿主免疫参与代谢紊乱的控制或发展,与全身慢性炎症相关[44]。肥胖个体PD的发展和严重程度的增加导致肠道菌群失调,这与P.gingivalis等机会致病菌的全身播散密切相关。肠道微生物群可以消化某些人类因缺乏酶而无法消化的膳食多糖,如β-葡萄糖苷酶、木聚糖酶、内切葡聚糖酶等[45],此过程产生短链脂肪酸(SCFA)。SCFA容易被吸收,促进脂肪生成相关分子途径,抑制脂蛋白脂肪酶(LPL)的活性,但这会导致甘油三酯在宿主脂肪细胞中的积累[46]。动物实验表明,给小鼠喂食高脂饮食后,持续性口服P.gingivalis会导致小鼠肥胖、糖耐量受损和加重肝脏脂肪变性[47]。P.gingivalis还能改变棕色脂肪组织(BAT)的内分泌功能,P.gingivalis注射可能导致BAT的葡萄糖稳态和胰岛素敏感度下降[48]。该过程与脂联素相关,又称脂肪连接蛋白,是一种脂肪细胞产生的激素,可改善血脂异常和胰岛素抵抗。研究显示,在使用P.gingivalis-LPS处理的脂肪细胞中,脂联素的水平明显下调[49]。此外,尽管P.gingivalis感染对雌性小鼠的血浆甘油三酯水平没有明显改变,但小鼠肝脏甘油三酯合成的基因表达下降,此结果可能会影响胎儿的生长发育。P.gingivalis感染导致母鼠妊娠期体重增加,胎儿体重降低[39,50]。P.gingivalis及其独毒力因子主要通过引起肠道菌群失调及改变脂肪细胞内分泌功能来促进个体肥胖。尽管营养不均衡和肠道微生物群失调本身会诱发肥胖症,伴随的PD会增强胰岛素抵抗,P.gingivalis的LPS则是PD的辅助剂。此外,与肥胖相关的某些代谢性疾病如“代谢综合征”则可能进一步加剧PD。
7 小结与展望
本文总结了P.gingivalis对多种疾病中的作用,更具体的致病机制仍需深刻探讨。P.gingivalis不仅是牙周炎的主要病原体,还能随着血液播散到全身其他脏器,直接或间接引起各种损伤。不良的口腔卫生会导致PD,并可能间接增加患AD的风险;如果AD患者在保持适当口腔卫生方面存在障碍,则会增加患PD的风险,提示保持良好的口腔健康可能成为预防AD的一种措施。对于不良妊娠结局,目前缺乏证据表明产前口腔保健可以改善其影响。值得注意的是,世界卫生组织于2007年提出应促进口腔健康,将其作为健康生活方式中不可或缺的一部分。通过改善口腔卫生及牙周状况,有望降低P.gingivalis的负荷,在一定程度上减少这些疾病对全身状况产生的不良影响。
猜你喜欢
现代临床医学(2021年4期)2021-07-31
中国眼镜科技杂志(2019年9期)2019-11-11
安徽医科大学学报(2016年12期)2017-01-15
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国病理生理杂志(2015年8期)2015-12-21
安徽医科大学学报(2015年9期)2015-12-16
医学研究杂志(2015年12期)2015-06-10
医学研究杂志(2015年11期)2015-06-10
云南中医学院学报(2014年5期)2014-07-31
现代检验医学杂志(2014年1期)2014-02-06
