
女性生殖系统血管肌纤维母细胞瘤7例临床病理分析
2023-03-10 02:18文保钢
重庆医学 2023年4期
文保钢,李 林,陈 锐,王 冬
(重庆大学附属肿瘤医院/肿瘤转移与个体化诊疗转化研究重庆市重点实验室,重庆 400030)
血管肌纤维母细胞瘤(AMF)是一种罕见的良性间质细胞肿瘤[1],好发于女性生殖系统,尤其是外阴部位,其次为阴道、宫颈、输卵管、阔韧带等,也可见于男性生殖道的腹股沟、阴囊及阴茎等[2-4]。目前,国内外相关病例报道较罕见。本研究回顾性分析重庆大学附属肿瘤医院妇科肿瘤中心2010年1月至2021年12月收治的7例女性生殖系统AMF临床病理资料,结合文献复习,探讨其临床病理特征、诊断、治疗及预后。现报道如下。
1 资料与方法
1.1 一般资料
重庆大学附属肿瘤医院妇科肿瘤中心自2010年1月至2021年12月共收治AMF患者7例,其中外阴5例,宫颈1例,臀部1例,均有完整的临床病理资料,所有患者病理切片均经2名高年资病理科医生复查核实。
1.2 方法
结合相关文献复习并回顾性分析7例AMF患者的临床病理资料。
2 结 果
2.1 临床特点
患者发病年龄为36~55岁,中位发病年龄46岁,其中绝经后发病者3例。AMF以发现外阴、阴道、臀部肿物为主要症状,病程6~36个月,平均14个月。妇科检查提示肿物主要位于大阴唇4例(2例位于左侧,2例位于右侧),左侧阴阜1例,宫颈1例,右侧臀部(上隆起至右侧大阴唇,下隆起至坐骨结节附近)1例。肿物最大直径为1~8 cm,平均4 cm,其中5例呈质硬的实性肿物,2例呈囊性,肿物均可活动,边界清楚。术前检查肿瘤标志物包括糖类抗原125(CA125)、CA19-9、癌胚抗原(CEA)、甲胎蛋白(AFP)、鳞癌抗原(SCC)等,数值均在正常范围。超声检查均提示肿物异常回声,边界清楚,3例考虑囊肿,1例考虑血管瘤,1例考虑肿大淋巴结,1例考虑肌瘤,1例考虑脂肪瘤。术前4例患者诊断为外阴肿物,1例诊断为外阴囊肿,1例诊断为臀部肿物,1例诊断为宫颈肌瘤(表1)。
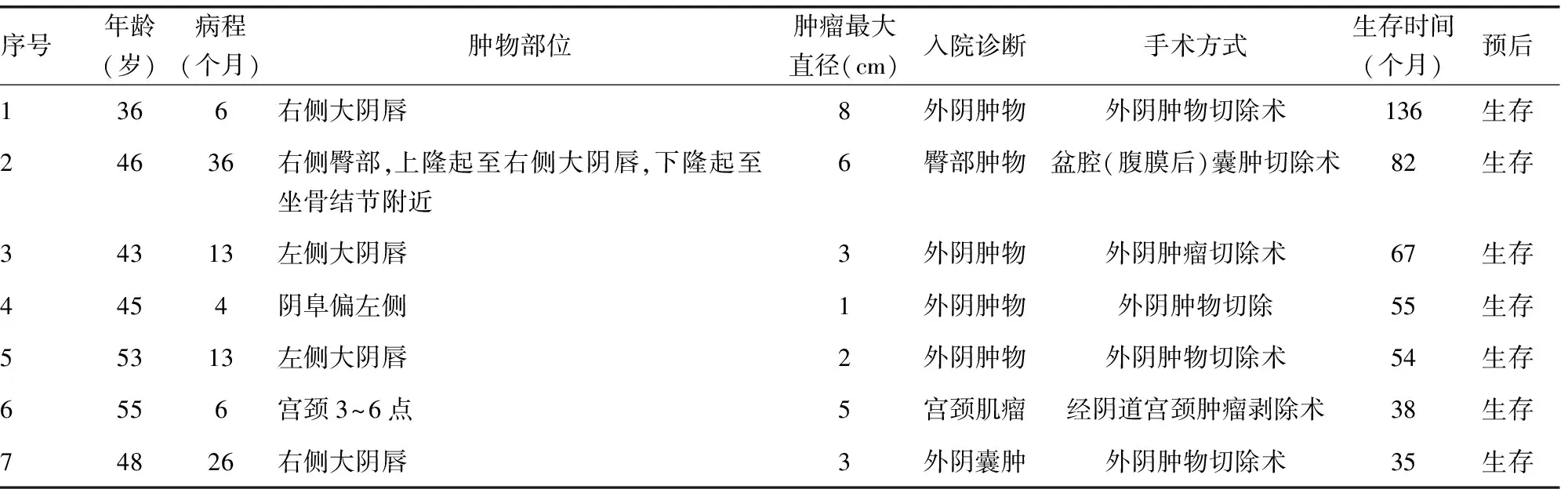
表1 AMF患者临床资料
2.2 病理及免疫组织化学特点
肿物大体标本提示,肿物质中或质软,包膜多完整;镜下见肿瘤边界清楚,细胞密集区和细胞稀疏水肿区交替分布,可见丰富薄壁血管(图1A);梭形细胞包绕血管周围(图1B)。6例患者术后行病理及免疫组织化学检测,波形蛋白(vimentin)阳性6例,雌激素受体(ER)阳性6例(图1C),孕激素受体(PR)阳性5例(图1D),CD34阳性5例(图1E),结蛋白(desmin)阳性3例(图1E),S-100、细胞角蛋白(CK)等均为阴性,Ki-67阳性5例(图1F),且均≤10%。术后病理均诊断为AMF,2例部分区域兼类似侵袭性血管黏液瘤改变,1例部分区域细胞伴轻度异型。见表2。

表2 AMF患者病理及免疫组织化学结果
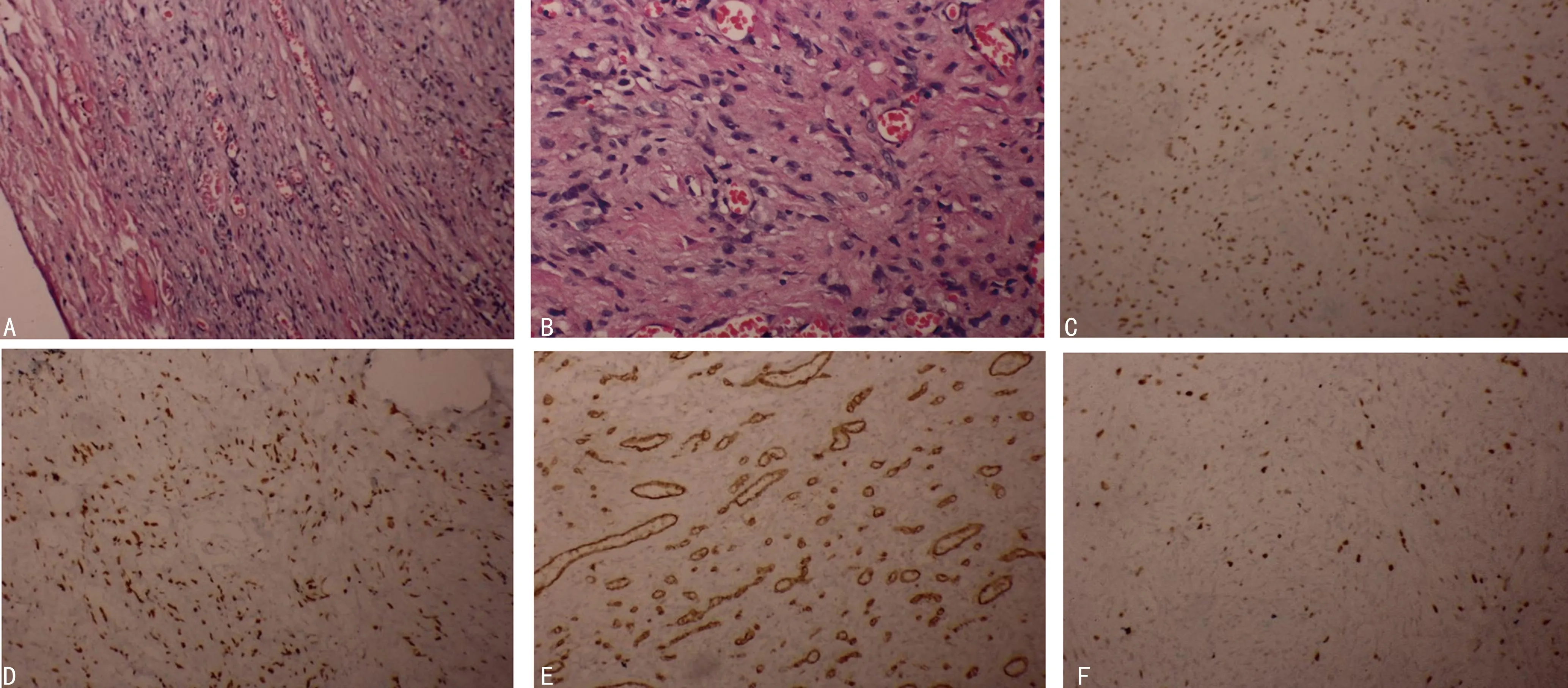
A:苏木精-伊红(HE)染色(100×),肿瘤边界清楚,由交替分布的细胞密集区和细胞稀疏水肿区构成,可见丰富薄壁血管。B:HE染色(200×),梭形细胞包绕血管周围。C:链霉亲和素-过氧化物酶(SP)法(100×),瘤细胞核ER强阳性。D:SP法(100×),瘤细胞核PR强阳性。E:SP法(100×),瘤细胞CD34呈弱阳性。F:SP法(100×),瘤细胞Ki-67约10%阳性。
2.3 治疗
7例患者均行肿物切除术,其中2号患者术前影像学提示肿物位于右侧坐骨肛门窝、右臀部及右会阴部,最大截面为9.6 cm×5.6 cm×5.3 cm,边界不清,病变突破会阴浅深筋膜侵入盆腔,右侧肛提肌疑受侵,宫颈、直肠受压向左移位。术中行尾骨尖至右侧坐骨结节的弧形切口,切开皮下组织后,见囊肿与皮下组织关系密切。沿着囊肿的表面,用超声刀向外侧分离臀部脂肪与囊肿的粘连,继续向内侧、上方及盆腔方向游离囊肿。分离过程中,发现囊肿与阴道、右侧坐骨结节、肛提肌等关系紧密,完整游离并切除肿物。
2.4 预后
随访截至2022年5月,全部病例均进行了电话随诊或就诊复查,中位随诊时间67个月(47~148个月),患者均存活,无1例复发。
3 讨 论
AMF总体发生率低,好发于30~50岁成年女性,男性罕见。目前多为小病例报道,有学者汇总分析了71例外阴AMF,患者的平均年龄为45岁(17~86岁);临床症状主要是逐渐增大的无痛性肿物;平均病程29个月(2个月至10年);52%位于左侧外阴,48%位于右侧外阴;肿物平均直径5.9 cm(0.5~30.0 cm)[1]。AMF超声可表现为高回声或低回声,界限清楚;增强计算机断层扫描显示肿瘤呈中度增强;但均无特异性[5-6]。术前易误诊为巴氏腺囊肿、脓肿和脂肪瘤等[1,7]。本组7例患者中位发病年龄46岁,临床表现为局部发现无痛性肿物,其中5例位于外阴,1例位于宫颈,1例位于臀部,发生于外阴部位的主要位于大阴唇。本组病例肿物平均直径为4 cm,考虑与平均病程较短有关。超声检查对确定肿物位置、大小及边界有帮助,本组病例术前彩色多普勒超声检查考虑囊肿有3例、血管瘤1例、肿大淋巴结1例、肌瘤1例、脂肪瘤1例。术前诊断困难,需术后病理及免疫组织化学确诊。
AMF病因不明。王海燕等[8]认为该肿瘤可能是因为血管内皮周围干细胞受到局部损伤、炎症刺激等作用,向肌纤维母细胞分化,而后者在雌激素等多种因素刺激下出现异常增殖,最终形成肿瘤。本组6例患者术后行病理检查,6例ER阳性,5例PR阳性,支持“雌激素刺激学说”。AMF与其他部位特异性间充质肿瘤表型不同,可能有共同的肿瘤发生机制[9]。WANG等[10]对1例外阴AMF进行全外显子测序分析,显示有基因组的改变,这可能是导致AMF发生的原因之一。
AMF肿物大多呈卵圆形或圆形,部分呈息肉状,大多界限清楚,切面实性,部分可呈囊实性,局灶伴黏液样或胶冻样[11]。本组2例为囊实性,余均为实性,6号患者(1例)呈息肉状。镜下见肿瘤组织边界清楚,薄壁血管丰富;细胞密集区和稀疏水肿区交替出现。瘤细胞的细胞质半透明,呈淡嗜伊红色,核偏位,核仁不明显,无细胞异型性(图1)。AMF与其他软组织肿瘤组织形态相似,较难区分,因此免疫组织化学检测具有重要价值。AMF细胞表达vimentin和desmin,多不表达S-100、CK;部分表达ER、PR、CD34等[1,12]。本组6例瘤细胞均表达vimentin及ER,不表达CK与S-100,Ki-67阳性指数均≤10%。
AMF鉴别诊断尤为重要,需要与以下肿瘤相鉴别。(1)侵袭性血管黏液瘤。侵袭性血管黏液瘤一般发生于25~50岁女性;临床上表现为相对局限、体积较大、缓慢生长的多叶状、息肉状或黏液样肿物,并延伸到周围组织,肿瘤大小从数厘米至20 cm或更大者均有,边界不清楚;镜下血管大小不一、管壁厚薄不等;肿瘤细胞呈星状、梭形稀疏均匀分布;间质黏液样变性明显[11]。肿瘤细胞弥漫性表达desimin;60%~70%表达高迁移率族蛋白A2[13]。与侵袭性血管黏液瘤相比,AMF通常表现为生长缓慢、无痛的皮下肿物,多数直径<5 cm,极少数为大的带蒂肿物;边界清楚、通常无包膜[11];镜下具有更高的细胞数量、更多的血管数量和更频繁的梭形细胞[1]。该病完整切除困难,局部复发率高,属于低度恶性肿瘤。本组2例患者局灶伴有侵袭性血管黏液瘤改变,确保完整切除是避免复发的关键。(2)浅表性血管黏液瘤。浅表性血管黏液瘤多见于成年男性躯干、头颈及下肢,很少发生于生殖器;呈结节状或息肉分叶状肿物,界限尚清但无包膜;由散在的短梭形或星状纤维母细胞组成;有大量黏液样基质,可见薄壁、狭长的血管,并有少量的中性粒细胞浸润;免疫组织化学desmin为阴性,平滑肌肌动蛋白偶有阳性;30%~40%患者因切除不全致局部复发[14]。相较于浅表性血管黏液瘤,AMF更常见于女性外阴及阴道;镜下具有更丰富嗜酸性细胞质的上皮样或浆细胞样细胞;可单纯切除,无复发,无转移[14]。
目前,AMF无相关有效药物治疗报道,包块生长增大后压迫周围组织器官会出现相应症状,首选手术治疗,建议行保留一定无瘤边界的完整肿物切除术[15]。术中彩色多普勒超声对判断手术切除范围有一定帮助,术后病理确诊后随访观察即可。本病预后良好,建议长期随访,警惕恶变可能。
猜你喜欢
伴侣(2022年2期)2022-03-30
世界最新医学信息文摘(2021年12期)2021-06-09
中国民间疗法(2021年6期)2021-06-09
幸福家庭(2021年1期)2021-03-08
中国临床医学影像杂志(2019年5期)2019-08-27
中国临床医学影像杂志(2019年2期)2019-04-25
妈妈宝宝(2017年3期)2017-02-21
现代检验医学杂志(2016年3期)2016-11-15
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
中国卫生标准管理(2015年1期)2016-01-14
