
Comparative Proteomic Study on The Lesional and Non-lesional Epidermis From Vitiligo Patients*
2023-02-26 07:52AilikemuTuerxunCHENXiuLanAiniwaerTalifuLINaWANGJiFengCAITanXiGUOXiaoJingDINGXiangXIEZhenShengNIULiLiZHANGMengMengGhulamAbbasHajiAkberAisaYANGFuQuan
生物化学与生物物理进展 2023年2期
Ailikemu Tuerxun ,CHEN Xiu-Lan ,Ainiwaer Talifu ,LI Na ,WANG Ji-Feng ,CAI Tan-Xi ,GUO Xiao-Jing,DING Xiang,XIE Zhen-Sheng,NIU Li-Li,ZHANG Meng-Meng,Ghulam Abbas,Haji Akber Aisa,YANG Fu-Quan**
(1)Key Laboratory of Plant Resources and Chemistry of Arid Zone,Xinjiang Technical Institute of Physics and Chemistry,Chinese Academy of Sciences,Urumqi 830011,China;2)University of Chinese Academy of Sciences,Beijing 100049,China; 3)Laboratory of Protein and Peptide Pharmaceuticals and Laboratory of Proteomics,Institute of Biophysics,Chinese Academy of Sciences,Beijing 100101,China; 4)Hospital of Xinjiang Traditional Uyghur Medicine,Urumqi 830049,China)
Abstract Objective The comparative proteomic study on the paired lesional epidermis (LE) and non-lesional epidermis (NLE) from vitiligo patients to identify the differentially expressed proteins (DEPs) between LE and NLE,and to further explore the molecular mechanism of pathogenesis of vitiligo.Methods Firstly,the in solution digestion condition for proteins from epidermis were optimized to sequential tandem digestion with Lys-C and trypsin.Secondly,tandem mass tay (TMT) based quantitative proteomic strategy was performed to compare the proteome profile of the paired LE and NLE from three stable non-segmental vitiligo subjects,and differential expressed proteins (DEPs) were identified.At last,the functional enrichment analysis was performed via bioinformatics tool and database (GO,KEGG,STRING,GSEA).Results The optimal sequential tandem digestion condition was the combination of Lys-C (enzyme∶substrate,1∶100) and trypsin (enzyme∶substrate,1∶50).A total of 4 496 proteins were identified,and of which 181 were DEPs between LE and NLE from vitiligo patients.Bioinformatics analysis showed that DEPs were mainly related with metabolism,immunity,redox and cell adhesion.Among them,the 119 up-regulated proteins are mainly involved in the processes of keratinization,transcription,oxidative stress,and proteolysis.The 62 down-regulated proteins are mainly involved in intracellular transport,glutathione metabolism and actin filament capping.Conclusion The comparative proteomic study revealed that there were functional differences in keratinization,immunity,lipid metabolism and redox between LE and NLE in vitiligo patients.PRDX1,PRDX2,EEF2,ITGB1,SPTBN2,ANXA1 and PFKL were found as the key proteins to disfunction of LE.
Key words vitiligo,epidermal proteomics,TMT labeling,keratinization,oxidative stress,lipid metabolism
Vitiligo is a common depigmenting skin disorder,characterized by patchy skin bordered milky-white depigmentation accompanied by melanocytes disappearance from lesional skin.The worldwide prevalence is between 0.5%-2% without any significant difference in sex,ethnicity,and geographic region.Vitiligo is not life-threatening but influences the quality of life[1-2].Vitiligo is a multifactorial disorder and there are many hypotheses about the pathogenesis of vitiligo,including genetics,autoimmune,oxidative stress,melanocyte adhesion,and neural factors[3-4].However,none of these hypotheses clearly explained the mechanism of the pathogenesis of vitiligo.
Quantitative proteomics is an excellent largescale screening tool for the discovery of the differential expressed proteins (DEPs) and thus could provide a better understanding of the entire protein network[5-7].However,only the two-dimensional gel electrophoresis (2D-GE) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) based proteomic strategy has been used to identify the DEPs in the serum of vitiligo patients[8-9]in the previous proteomics studies on vitiligo.So far,proteomic study on lesional epidermis of vitiligo patients has not been reported.
In this study,in order to identify the features of pathological changes of the lesional epidermis deprived of melanocytes in vitiligo patients,and to explore the molecular mechanism of pathogenesis of vitiligo,TMT-based quantitative proteomic strategy was used to compare the proteome profile of the paired LE and NLE lesional epidermis collected from three stable non-segmental vitiligo patients to identify the DEPs (Figure 1).
1 Materials and methods
1.1 Sample collection
Vitiligo was diagnosed based on the patient’s medical history and typical clinical features (lesions location,size,distribution) by experienced dermatologists accompanied by the Wood’s lamp.The patients with non-segmental vitiligo (NSV) were chosen as participants.The criteria for the patients chosen in this study was based on two conditions: (1) no expansion on preexisting lesions,absence of new lesions and without Koebner phenomenon for at least six months;(2) no external or systemic treatment of vitiligo for at least three months.All participants signed an informed written consent form before the study was initiated.BFY-10 dermis-epidermis separator (negative pressure ranging from 40 to 60 kPa,suction cup with inner diameter of 0.8 cm) was used to induce suction blister.Epidermis was obtained from the center of lesional skin and the paired non-lesional skin (at least 15 cm away from the border of the lesional skin) from the same patient.The six paired epidermis samples from three patients were collected in the Department of Dermatology,Hospital of Xinjiang Traditional Uyghur Medicine.
1.2 Protein extraction from the epidermis
Epidermis samples were washed twice with 500 µl ice-cold phosphate-buffered saline (PBS) and homogenized (OMNI TH-02,US) in 300 µl lysis buffer (6 mol/L urea,2 mol/L thiourea,100 mmol/L HEPES,pH 8.5) and 1% protease inhibitor (Roche,cocktail) for 2 min on ice followed by sonication (Scientz-IID,China) for 10 min on ice-water mixture.The lysate was centrifuged at 20 000×gat 4℃ (Eppendorf,centrifuge 5424 R) for 10 min.The supernatant was collected and stored at -80℃ for further use.The Bradford assay was used to determine the total protein concentration.

Fig.1 Schematic illustration of workflow of proteomic analysis
1.3 In solution digestion and desalting
All details and data about the optimization of in-solution digestion conditions for epidermis proteins are in the supplementary materials (Figure S1,File S1,Table S1).50 µg of extracted proteins was taken from each sample and reduced in 10 mmol/L dithiothreitol (DTT) at 37℃ for 1 h,while alkylated in 20 mmol/L iodoacetamide (IAM) for 30 min in the dark,at room temperature.After that,Lys-C (Wako,Japan) was added into the sample solution at a ratio (enzyme/substrate) of 1∶100 (w/w) and incubated at 37℃ for 3 h.Then,the sample solution was diluted for five folds with 50 mmol/L NH4HCO3,and trypsin (Promega) was added at a ratio (enzyme/substrate) of 1∶50 (w/w) and incubated at 37℃ overnight.Formic acid (FA) was then added into the sample solution at a final concentration of 0.5% to stop the digestion.The tryptic peptide samples were desalted with HLB C18 columns (Waters,MA,USA),and lyophilized with vacuum centrifugation (LABCONCO).
1.4 TMT 6-plex labeling
All the peptide samples were dissolved in 100 mmol/L TEAB buffer,and 35 µg peptides were taken from each sample solution and labeled with TMT 6-plex reagent (RF232465,Thermo Fisher Scientific,USA) according to the manufacturer’s protocol.TMT126-128 reagents were used to label the NLE samples,and TMT129-131 reagents were used to label the LE samples.After labeling,the TMT 6-plex-labeled peptide samples were pooled,desalted,and dried for the next high pH reversed-phase fractionation.
1.5 High pH reversed-phase fractionation
High pH revered-phase (RP) fractionation was carried out to reduce sample complexity and increase the depth of proteome identification as described previously[10].Briefly,the TMT 6-plex-labeled peptide samples were dissolved in buffer A (2% ACN/98% H2O,pH=10,adjusted with ammonium hydroxide) and loaded onto an XBridge C18 basic reversed-phase LC column (100 mm×2.1 mm,3.5 μm particles,Waters).Peptides were separated in a Rigol L-3000 LC system with a binary buffer system of buffer A and buffer B (98% ACN/2% H2O,pH=10) at a gradient (5%-8% B,0-5 min;8%-18% B,5-40 min;18%-32% B,40-62 min;32%-95% B,62-64 min;95%-95% B,64-68 min;95%-5% B,68-69 min;5%-5% B,69-76 min) at a flow rate of 0.2 ml/min).A total of 42 fractions were collected and combined into 12 fractions.These combined fractions were dried and desalted for LC-MS/MS analysis.
1.6 LC-MS/MS analysis
All peptide samples were analyzed on EASYnLC 1000 system (Thermo Fisher Scientific) coupled with a Q Exactive mass spectrometer (Thermo Fisher Scientific).All peptide samples were dissolved in 0.1% FA and loaded onto an in-house packed capillary RP trap column (100 μm ID×2 cm,Reprosil-Pur C18 AQ,5 μm,Dr.Maisch GmbH),and separated with a capillary RP C18 column (75 μm ID×20 cm,Reprosil-Pur C18 AQ,3 μm,Dr.Maisch GmbH) with buffer A (0.1% FA) and buffer B (100% ACN/0.1% FA) at a gradient (4%-12% B,0-5 min;12%-22% B,5-55 min;22%-32% B,55-67 min;32%-90% B,67-68 min;90%-95% B,68-75 min).The flow rate was set to 310 nl/min.Peptide elutates from the column were directly introduced into the mass spectrometer with a spray voltage of 2.0 kV and a capillary temperature of 320℃.
The mass spectrometer was operated in a datadependent (DDA) mode,MS1 scans at a resolution of 70 000 with an automatic gain control target of 3×106ions and a maximum ion injection time of 60 ms.The top 20 most abundant peaks were selected for fragmentation with an isolation window of 2m/zand were fragmented by higher-energy collisional dissociation with a normalized collision energy of 27%.Fragmentation spectra were acquired at a resolution of 17 500 with a target value of 5×104ions and a maximum ion injection time of 80 ms.To minimize peptide re-sequencing,dynamic exclusion was enabled within a time window of 40 s.
1.7 Database searching
Proteome Discoverer software (v2.2) with SEQUEST HT search engine was used for protein identification.Reporter ion intensity was used for protein quantification.MS/MS spectra were searched against Uniprot human-reviewed database (retrieved on December 21,2020) supplemented with known contaminants.Peptide mass and fragment mass tolerances were set at 10 ppm and 0.02 u,respectively,and a maximum of 2 miss-cleavages were allowed.Cysteine carbamidomethylation,TMT 6-plex labeled lysine (K) and N-terminus of peptides were set as fixed modifications,and N-terminal acetylation on proteins,methionine oxidation were set as variable modifications.Peptide identifications were filtered at a 1% false discovery rate and only high confident peptides were accepted for further analysis.
1.8 Bioinformatics analysis
High confident quantified proteins were used for further analysis.Statistical analysis was performed with paired two-sample Student’st-tests.Proteins with fold change (FC) (LE compared to NLE) of more than 1.2 (up-regulated) or less than 0.83 (downregulated) withPvalue<0.05 were considered as DEPs.Network analysis of DEPs was performed with DAVID Bioinformatics Resources 6.8 (the total genes in the genome as a background) and STRING (Version 11.5).Gene set enrichment analysis was performed with GSEA 4.1.0 and the terms with false discovery rate (FDR) < 0.05 were visualized with Cytoscape (Version: 3.8.2).
2 Results
2.1 Optimization of protein digestion condition
The protein digestion condition was optimized by comparing four different digestion protocols with combination of Lys-C and trypsin at different ratio of enzyme/substrate.More suitable length of peptides with less missed cleavage under the Lys-C and trypsin sequential digestion protocols (protocols 1,2,3) than trypsin digestion alone (protocol 4).The optimized digestion condition protocol 1 (Lys-C 1:100+trypsin 1:50) could achieve the best identification results with 18%-36.4% increase of more proteins identified compared with other conditions (Figure S2).
2.2 Identification of DEPs
TMT-based quantitative proteomic technology was utilized to compare protein expression between LE and NLE of vitiligo patients.A total of 4 496 proteins were identified from TMT-labeled samples.3 736 proteins,which had quantification information in all the six channels,were selected for further analysis.181 proteins were regarded as DEPs,with 119 up-regulated and 62 down-regulated (Figure 2a).Principal component analysis (PCA) of these DEPs showed a clear separation with distinct features between LEs and NLEs (Figure 2b).

Fig.2 Comparison of LE and NLE on proteins quantified in epidermis proteomics
2.3 DEPs enrichment analysis
Using GO analysis in DAVID Bioinformatics Resources,functional enrichment of the DEPs was categorized with biological process (BP),cellular component (CC),and molecular function (MF).As shown in Figure 3,for the up-regulated proteins,the top 10 significantly enriched BP were associated with the proteolysis,neutrophil degranulation,translation,cornification (keratinization),cellular response to oxidative stress,and cadmium ion.The CC analysis showed these proteins were enriched in extracellular parts including exosome and extracellular region,followed by cytosol,granular lumen,focal adhesion,lysosome,and endoplasmic reticulum.For MF analysis,these proteins were associated with peptidase and inhibitor activity,followed by oxidoreductase activity,binding,and transferase activity.
For down-regulated proteins,the enriched BP terms included neutrophil degranulation,intracellular transport,actin filament capping,glutathione metabolic process,and response to calcium ion.The enriched CC terms were extracellular region with exosome focal adhesion and membrane,followed by the cortical actin cytoskeleton,granular lumen,and neuronal cell body.The MF predominantly enriched in binding functions include actin binding,phospholipid,collagen,cadherin,and glutathione binding,followed by phospholipase inhibitor activity and oxidoreductase activity.
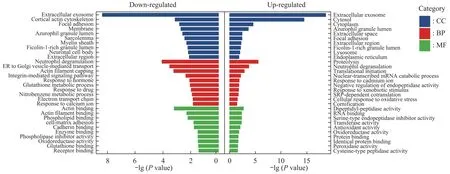
Fig.3 GO analysis of DEPs in epidermis of vitiligo
Interestingly,both of the up-regulated and downregulated proteins shared several common enrichment terms,such as the neutrophil degranulation term for BP,extracellular region,exosome,adhesion,and granular lumen terms for CC,and oxidoreductase activity for MF.
To uncover DEPs-associated pathways,KEGG pathway enrichment analysis was conducted with DAVID.As shown in Figure 4,DEPs associated with the cargo concentration in the ER,oxidatively deaminated to aldehydes by monoamine oxidase A(MAOA) and monoamine oxidase B (MAOB),eukaryotic translation termination,amino acid synthesis,vesicle biogenesis with transport,detoxification of reactive oxygen species (ROS),and formyl peptides binding.

Fig.4 KEGG pathway analysis of DEPs in epidermis of vitiligo
2.4 Protein-protein interaction(PPI)network construction
PPI network of DEPs was performed by STRING online database (Version 11.5).Each node represents a protein,color in nodes represents different GO functions (red for metabolic process,green for oxidation-reduction process,purple for immune system process,yellow for cell adhesion).The edge represents protein-protein interaction,different colors of edges indicate different functional and physical protein associations.PPI network construction carried out under the condition of minimum required interaction score was 0.4 (medium confidence).As shown in Figure 5,180 nodes (only nodes with interactions were shown) with 102 edges were found (PPI enrichmentPvalue: < 4.16×10-8).Functional enrichments analysis shows that among the 181 DEPs there are 119 proteins involved in the metabolic process,42 proteins involved in immune system process,27 proteins take part in the oxidationreduction process,and 15 proteins involve in cell adhesion.The multicolored nodes and edges obviously indicate that there is a close relationship between the different protein clusters.These multicolored proteins with high interaction scores (≥0.7) need further analysis.These proteins include PRDX1,PRDX2,EEF2,ITGB1,SPTBN2,ANXA1,and PFKL,and may play an important role in the pathogenesis of vitiligo.
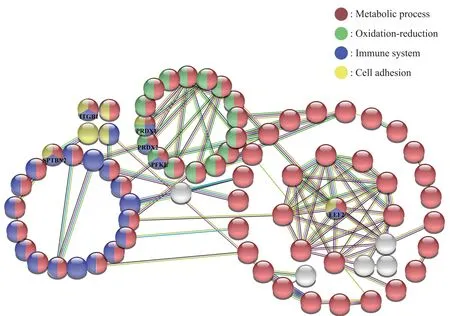
Fig.5 Protein-protein interaction network of DEPs
2.5 Gene set enrichment analysis
We also analyzed the gene set enrichment network of all quantified proteins (including changed and unchanged) by GSEA (Gene set enrichment analysis 4.1.0),and analysis was visualized with Cytoscape (Version: 3.8.2).Applying the thresholds (Pvalue<0.05,FDR<5%) as cutoff criteria.As shown in Figure 6,a total of 315 gene sets (nodes represent gene sets) were found and most of the down-regulated gene-sets clustered to immune response,followed by cell adhesion,migration,junction,wound healing,endocytosis,and lipid transport,whereas all the clusters but lipid transport above were related with each other (edges represent overlap).The upregulated gene sets were involved in macromolecules (including DNA,RNA,protein) metabolism,keratinization,and oxidant detoxification.

Fig.6 GSEA enrichment map of vitiligo LE vs NLE
3 Discussion
3.1 Enhanced keratinization(cornification)in LE
Physiological skin protection against UV radiation damage includes stimulation of melanin synthesis,enhanced keratinization,and increasing the antioxidant activity[11].
In the current study,GO and GSEA analysis shows that keratinization is enhanced in LE compared to the NLE.GO analysis enriched keratinization associated proteins include Keratin76 (KRT76),Keratin16 (KRT16),Caspase-14 (CASP14),and Lipase member N (LIPN),while KRT76 is the highest expressed protein in LE.KRT76 is a type II cytoskeletal intermediate filament protein that protects epithelial cells from both mechanical and nonmechanical stresses[12].Another significantly up-regulated keratin is KRT16,which is reported to contribute to hyperproliferation,cell adhesion,and migration[13].CASP14 is a different caspase family member that is activated in the normal keratinization process without activating apoptosis[14].LIPN is involved in the last step of keratinocyte differentiation and lipid metabolism[15].In all,we speculated that these four proteins participate in the process of keratinization and play important role in protecting the skin from the UV radiation.
3.2 Lipid metabolism in LE
STRING online database analysis showed that most of DEPs were involved in the metabolic process,and 25 DEPs were lipid-associated proteins.GSEA analysis also showed a cluster of gene sets associated with lipid metabolism.So,we investigated these lipidassociated proteins in detail.Fatty acid-binding protein 5 (FABP5) is one of the significantly up-regulated proteins in LE.FABP5 is a class of cytosolic lipid-binding protein that controls hydrophobic lipids transport and metabolism[16-17].Overexpression of FABP5 enhances lipid metabolism,mediating uptake of fatty acids and reducing the levels of cellular free fatty acid[18].Up-regulation of FABP5 may imply that LE has greater fatty acid uptake and enhanced lipid metabolism than NLE.Apolipoprotein C1 (APOC1) inhibits lipoprotein binding with low-density lipoprotein (LDL) receptor,very-low-density lipoprotein (VLDL) receptor,and LDL receptor-related protein (LRP)[19].Only basal layer keratinocyte expresses the LDL receptors[20-21].APOC1 affects the lipid composition of the epidermis[22].In our study,we found APOC1 downregulated in LE,indicating more LDL uptake into basal keratinocytes.Besides,we found that Acetyl-CoA acetyltransferase (ACAT1) is also downregulated in LE,resulting in more free cholesterol and fewer cholesterol esters in keratinocytes.
From analysis above,we speculated that there is increased lipid uptake and metabolism in LE vitiligo than NLE vitiligo.The lipid metabolic difference in LE might be essential for the keratinization that supplies the energy source for proliferation,and maybe the change in the epidermis lipid composition by APOC1 offer more UV protection than NLE.
3.3 Immunity in LE
GSEA analysis and STRING PPI analysis indicated that another group of proteins related with vitiligo was immunity The highest enriched gene sets function in GSEA analysis was immunity.42 DEPs involved in the immune system process from STRING PPI analysis,among which 4 T-cell enriched proteins (overlapped with TOP 100 T-cell enriched proteins[23]) were identified,including POSTN,APOH,F5,and ARG1.POSTN,APOH and F5 were down-regulated which may indicate decreased amount of T-cell in LE.ARG1 is implicated in immunosuppressive processes,up-regulated ARG1 in LE suggests less immunity in LE[24-27].Moreover,the elevated ARG1 level stimulates keratinocyte proliferation[27-28].
In all,we assume that LE in stable vitiligo exhibits less immune activity than NLE.That is probably the cause of the diminished melanocytes in LE,as melanocytes are the source of inflammation,apoptosis,and recruited immune cells.
3.4 Amine oxidases(MAOs)in LE and NLE
KEGG pathway analysis shows that DEPs are involved in deaminated aldehydes by MAOA and MAOB.MAOs (MAOA and MAOB) are involved in the metabolism of dopamine,therefore MAOs closely are associated with neurodegenerative disorders[29-31].In addition,dopamine and melanin synthesis share the same preprocess[32].In our study,we found downregulated MAOA and up-regulated MAOB in LE,indicating that vitiligo pathogenesis is associated with neurotransmitters.
Nerve endings and keratinocytes release noradrenaline into the epidermal microenvironment that inhibits tyrosinase (vital enzyme in melanin synthesis) activity in epidermal melanocytes[33].In addition,MAOA especially degrades noradrenaline[34].The previous study showed that the expression levels of MAOs was lower in lesional skin than non-lesional in active vitiligo[32].In our study,we found down-regulated MAOA in LE.These findings indicate that down-regulated MAOA may be one of the reasons for disappeared melanin in LE.
In conclusion,down-regulated MAOA and upregulated MAOB in LE could be the promising marker of stable vitiligo and could also be one of the reasons for the disappearance of melanin in LE.Probably the evidence for vitiligo pathogenesis associated with neurotransmitters.
3.5 Oxidoreduction in LE
All of GO,KEGG,STRING,and GSEA analysis shows an enriched function on the oxidoreduction system.An abnormal oxidoreduction system is responsible for oxidative stress.Oxidative stress is caused by an imbalance between the reactive oxygen species (ROS) formation and the antioxidant ability to reduce these reactive intermediates[35].Oxidative stress has been demonstrated to play a major role in the onset and progression of vitiligo[36-37].Antioxidant activity is one of the important physiological skin protection against UV radiation[11].The epidermis of vitiligo patients exhibits a high level of ROS and reduced antioxidant capacity[36,38-39].In this study,we identified 33 antioxidants-related proteins some wellknown antioxidants (SOD,CAT,HO,GPXs) displayed no expression changes in LE and NLE,however,several antioxidant-related proteins were upregulated (KEAP1,GLRX,PRDX1,PRDX2,TXNDC17,TXNDC5) and down-regulated (GSR,GSTM2,GSTM3).These proteins are an important regulator of the redox system,among which,GLRX,GSR,GSTM2,and GSTM3 are essential for the proper functioning the glutathione (GSH) that is a main non-enzymatic thiol-containing antioxidant in mammalian cells[40].PRDX1,PRDX2,TXNDC17,and TXNDC5 are the other different antioxidant system that plays pivotal roles in regulating multiple cellular redox processes[41-42].Keap1 is a critical component of the Keap1-Nrf2 pathway,acting as antioxidants by sensing oxidative stress and regulating Nrf2 activity[43].Normally Keap1 over-expression leads the Nrf2 to be less translocated into nucleus and decreases the expression of target genes[44](target genes include PRDX1,GSR,GSTM2,GSTM3,GCLC,and HO1).Actually,differential expression levels of downstream target genes in our study indicate that these antioxidants may not only be targeted by Nrf2.
Furthermore,each of these differentially expressed antioxidants-related proteins are involved in many other processes apart from the antioxidation process.Several reports showed that GSR negatively correlates with UVA radiation[11,37,45].Overexpressed GLRX inhibits oxidative stress and apoptosis[46],performing a suppressive role in the immune response[47].In our study,STRING PPI online analysis shows that PRDX1 and PRDX2 are associated with immune and metabolic processes.So,we conclude that LE and NLE have different redox levels.
4 Conclusion
The comparative proteomic study revealed that there were functional differences in keratinization,immunity,lipid metabolism and redox between LE and NLE in vitiligo patients.PRDX1,PRDX2,EEF2,ITGB1,SPTBN2,ANXA1 and PFKL were found as the key proteins to disfunction of LE.
SupplementaryAvailable online (http://www.pibb.ac.cn or http://www.cnki.net):
PIBB_20220158_Fig S1.tif
PIBB_20220158_Fig S2.tif
PIBB_20220158_File S1.pdf
PIBB_20220158_Table S1.xlsx

- 生物化学与生物物理进展的其它文章
- Similarity of Binding Potentials Between Plant DUF538 and Animal Lipocalin: Cholesterol Binding Ability of DUF538*
- 脂联素在骨骼肌中的新功能及其与运动的关系*
- 基于脑电信号的癫痫发作预测特征及识别*
- Different Durations of Simulated Weightlessness Induced Depressive-like Behaviors in Rats*
- 洋山港宏病毒组分析揭示CRISPR-Cas系统病毒靶标序列的特异性*
- BE-dot:为单碱基编辑设计sgRNA及预测脱靶图谱的工具*