
液相色谱-串联质谱法测定畜禽肉和鸡蛋中氨基酸含量
2023-02-21 10:34:02李智周灿费华熙王素利郑连姬
食品与发酵工业 2023年3期
李智,周灿,费华熙,王素利,郑连姬*
1(重庆食品工业研究所有限公司,重庆,400042)2(中国轻工业联合会食品质量监督检测重庆站,重庆,400042)
氨基酸是蛋白质和酶的基本组成部分。氨基酸可按照氨基连在碳链上的不同位置而分为α-氨基酸、β-氨基酸、γ-氨基酸、ω-氨基酸。平常我们所知的经蛋白质水解后得到的氨基酸均为α-氨基酸,机体内常见氨基酸有20种,其中有8种氨基酸高等动物无法自主合成,包括赖氨酸、色氨酸、苯丙氨酸、蛋氨酸(又名甲硫氨酸)、苏氨酸、异亮氨酸、亮氨酸、缬氨酸,这些氨基酸必须通过不断摄取食物来获取。
氨基酸在体内重要的生理功能不仅是参与合成蛋白质和其他含氮化合物,还作为信号分子参与机体多种生理进程的调控,越来越多的学者证明氨基酸可作为信号分子参与细胞内信号传导过程,可以调控机体能量和糖脂代谢,最终导致机体整体代谢的改变[1]。如在厌氧状态下机体启动糖原分解时,谷氨酸和天冬氨酸对线粒体的氧化磷酸化和ATP合成进行调节[2];亮氨酸可以通过激活谷氨酸脱氢酶从而促进胰岛β细胞合成胰岛素[3]。
由于氨基酸分析在蛋白质化学、生物化学、食品科学、临床医学等领域的研究中起着重要的作用,因此,对氨基酸分析方法的研究与改进引起人们高度重视。目前报道的氨基酸检测方法有紫外分光光度法[4]、荧光光度法[5]、红外光谱法[6]、高效阳离子交换色谱法[7-8]、高效阴离子交换色谱-积分脉冲安培检测法[9]、高效液相色谱法[10]、毛细管电泳法(capillary electrophoresis,CE)[11]、气相色谱法[12]、气相色谱-质谱联用法[13]等。这些方法通常采用邻苯二甲醛[14]、异硫氰酸苯酯[15]、9-氯甲酸芴甲酯[16]、丹磺酰氯[17]、二硝基氟苯、1-氟-2,4-二硝基苯基-5-L-丙氨酰胺[18]、磺酰氯二甲胺偶氮苯[19]、6-氨基喹啉基-N-羟基琥珀酰亚胺基氨基甲酸酯[20]、2,4-二硝基氯苯[21]等衍生试剂进行衍生,这使得试剂消耗量大、检测步骤复杂、检测周期长。应用液相色谱串联质谱(liquid chromatography-tandem mass spectrometry, LC-MS/MS)检测氨基酸可以直接分析提取物中的氨基酸,无需采用衍生步骤[22-25]。本文通过对样品水解条件、质谱、色谱条件的优化,在电喷雾离子源(electron spray ionization, ESI)正离子扫描模式下检测,建立了液相色谱-串联质谱测定动物源性食品畜禽肉和鸡蛋中氨基酸的检测方法,并将检测结果与我国国家标准自动氨基酸测定方法进行了比较,以期建立一种适用于畜禽肉和鸡蛋中氨基酸检测的方法。
1 材料与方法
1.1 材料与试剂
氨基酸标准溶液(包括半胱氨酸、组氨酸、丝氨酸、精氨酸、丙氨酸、天冬氨酸、苏氨酸、谷氨酸、赖氨酸、脯氨酸、缬氨酸、甲硫氨酸、异亮氨酸、亮氨酸、苯丙氨酸、酪氨酸、甘氨酸),上海安谱实验科技股份有限公司;甲醇为色谱纯,河北百灵威超精细材料有限公司;盐酸、苯酚、甲酸、乙酸铵、柠檬酸钠等均为国产分析纯,重庆川东化工(集团)有限公司。猪肉、鸡蛋、鸡肉,市售。
1.2 仪器与设备
Agilent 6460 Triple Quad LC-MS/MS仪,美国安捷伦公司;TOPEX+微波消解仪,上海屹尧仪器科技发展有限公司;BGZ-240电热鼓风恒温箱,上海博迅实业有限公司医疗设备厂;RE-52A真空旋转蒸发仪,上海亚荣生化仪器厂;ZFK-040电热真空干燥箱,上海实验仪器有限公司。
1.3 试验方法
1.3.1 样品水解
1.3.1.1 酸水解
称取均匀试样2 g于水解管中,加入15 mL 6 mol/L HCl溶液,加入3~4滴苯酚,将水解管放入冷冻剂中,冷冻3~5 min,接到真空泵的抽气管上,抽真空(接近0 Pa),然后充入N2,重复抽真空-充入N23次后,在充N2状态下封口或拧紧螺丝盖。将已封口的水解管放在(110±1) ℃的电热鼓风恒温箱内,水解22 h后,取出,冷却至室温。打开水解管,将水解液过滤至50 mL容量瓶内,用少量水多次冲洗水解管,水洗液移入同一容量瓶(50 mL)内,最后用水定容至刻度,振荡混匀。准确吸取1.0 mL滤液移入到15 mL或25 mL试管内,用真空旋转蒸发仪在40~50 ℃加热环境下减压干燥,干燥后残留物用1~2 mL水溶解,再减压干燥,最后蒸干。用1.0~2.0 mL pH 2.2柠檬酸钠缓冲溶液加入到干燥后试管内溶解,振荡混匀后,吸取溶液通过0.22 μm滤膜后,备用。
1.3.1.2 高压水解
称取均匀试样0.5 g,置于50 mL消解管内,加入15 mL HCl溶液,加入2滴苯酚,于110 ℃鼓风干燥箱中消解6 h,取出冷却,将水解液全部转移至 50 mL容量瓶中,用一级水定容至刻度,摇匀、过滤,精密吸取1 mL于40~50 ℃加热环境下减压干燥,残留物用0.1%(体积分数,下同)甲酸溶液定容至刻度,摇匀,经0.22 μm的微孔滤膜过滤,备用。
1.3.1.3 微波水解
称取均匀试样0.5 g,置于100 mL消解管内,加入15 mL HCl溶液,加入2滴苯酚,于80 ℃消解炉内消解30 min,取出冷却,转移至微波消解仪上150 ℃,消解4 h,取出冷却,将水解液全部转移至50 mL容量瓶中,用一级水定容至刻度,摇匀、过滤,精密吸取1 mL于10 mL容量瓶中,置于真空干燥箱内,于50 ℃减压干燥(真空干燥箱内放入P2O5作为干燥剂),干燥后残留物用0.1%甲酸溶液定容至刻度,摇匀,经0.22 μm的微孔滤膜过滤,备用。
1.3.2 色谱条件
1.3.2.1 液相色谱条件
色谱柱:AthehaC8-WP(2.1 mm×150 mm,3 μm);流动相A为0.1%甲酸水溶液+10 mmol/L乙酸铵,流动相B为甲醇;流速0.3 mL/min;进样量10 μL;柱温25 ℃;梯度洗脱参数见表1。

表1 梯度洗脱程序Table 1 Program of gradient elution
1.3.2.2 质谱条件
离子源:ESI源;扫描方式:正离子模式;检测方式:多反应监测(multiple reaction monitoring, MRM);电喷雾电压:3 500 V;离子源温度:300 ℃;气流:5 L/min;雾化器压力:45 psi;鞘气温度:250 ℃;鞘气流:11 L/min;喷嘴电压:500 V。
1.3.3 方法学验证
检出限、精密度、回收率的测定。
1.4 数据统计与分析
每个实验重复3次,采用SPSS 19.0统计分析软件进行统计处理。
2 结果与分析
2.1 质谱条件优化
分别取5 μL质量浓度为1 μg/L氨基酸单标溶液,先后进行母离子和子离子扫描,获得稳定性好、信号强度高的碎片离子,通过MRM模式Fragmenton优化不同氨基酸的锥电压和CE电压。结果见表2。

表2 十七种氨基酸质谱参数Table 2 MS parameters for the 17 amino acids
2.2 色谱条件优化
在优化的质谱条件下,考察不同色谱柱和不同流动相对目标物质谱信号的影响。对比了3种色谱柱,ZORBAX Eclipse Plus C18(3.0 mm×150 mm,1.8 μm),ZORBAX SB-C18(2.1 mm×150 mm,1.8 μm),AthehaC8-WP(2.1 mm×150 mm,3 μm)。分别考察了甲醇-水、乙腈-水、甲醇-0.1%甲酸水溶液、乙腈-0.1%甲酸水溶液、甲醇-0.1%甲酸水溶液(10 mmol/L乙酸铵)、乙腈- 0.1%甲酸水溶液(10 mmol/L 乙酸铵)几种流动相对目标化合物的影响。结果显示,使用AthehaC8-WP(2.1 mm×150 mm,3 μm)色谱柱和甲醇-0.1%甲酸水溶液(10 mmol/L乙酸铵)体系,效果更好。甲醇较乙腈在离子响应及分离度上更好,而加入了甲酸可以明显提高17中氨基酸的离子化效率,乙酸铵能提高信号响应并能有效解决峰拖尾问题。在梯度洗脱条件下目标物出峰较均匀,峰形较好,分离度好,保留时间适中。17种氨基酸的总离子流图见图1。

1-Lys;2-His;3-Arg;4-Gly;5-Cys;6-Ser;7-Ala;8-Asp;9-Thr;10-Glu;11-Pro;12-Val;13-Met;14-LLE;15-Leu;16-Tyr;17-Phe图1 十七种氨基酸的总离子流图Fig.1 Total ion chromatogram of 17 amino acids
2.3 基质效应的研究
值得注意的是,动物源性食品在LC-MS/MS的分析方法中存在明显的基质效应,但是不含氨基酸的空白基质难以获取,无法采用配制基质匹配工作曲线的方法消除基质效应。另外氨基酸内标溶液价格异常昂贵,不利于成本控制。因此本实验增大样品提取液的稀释倍数以削弱甚至消除基质效应。实验过程中考察了将1.3.1中备用液稀释1、5、40、50倍(相当于称样后稀释500、2 500、20 000、25 000倍)时氨基酸的含量,实验结果见图2。随着稀释倍数的增大,各氨基酸的测定值也逐渐增大,稀释至20 000~25 000倍时,氨基酸含量基本不变,即稀释倍数500、2 500和20 000、25 000之间有显著性差异,稀释倍数为20 000和25 000之间无显著性差异。这说明各氨基酸在动物源性食品基质中呈现基质减弱效应,随着稀释倍数的增大,样液中基质含量相应减小,基质减弱效应也逐渐衰弱,稀释至20 000倍及以上时,样液中极微量的基质对氨基酸的影响基本可以忽略不计。因此在后续实验中将1.3.1中备用液稀释40倍(相当于称样后总稀释倍数为20 000倍),经0.22 μm的微孔滤膜过滤后上机测定。

图2 稀释倍数对基质效应的影响Fig.2 Effect of dilution ratios on matrix effect注:n=3,不同字母表示存在显著性差异(P<0.05)(下同)
2.4 样品水解条件优化
2.4.1 微波水解条件的优化
2.4.1.1 微波温度的优化
固定微波水解时间6 h,通过改变水解温度(130、140、150、160、170、180 ℃)考察氨基酸含量变化。结果表明,随着温度的升高,水解得到的氨基酸含量逐渐增大,150 ℃时氨基酸含量最大,高于150 ℃时部分氨基酸含量有所下降,因此选择150 ℃为最佳水解温度。
2.4.1.2 微波时间的优化
固定微波水解温度150 ℃,通过改变水解时间(2、3、4、5、6 h)考察氨基酸含量变化。结果表明,随着水解时间的增加,水解得到的氨基酸含量逐渐增大,4 h时氨基酸含量最大,时间>4 h时部分氨基酸含量略有下降,可能是因为个别氨基酸被破坏。因此选择4 h为最佳水解时间。
2.4.2 样品水解方法的确定
酸水解法蛋白质水解彻底,水解过程中不发生消旋化。缺点是该水解方法耗时长,步骤繁琐,容易对实验测定带来误差。酸水解法也是目前我国氨基酸检测国家标准中普遍采用的水解方法。高压水解法是以酸水解为基础,解决了酸水解方法耗时长、步骤繁琐的问题,一般6 h可以完成。微波水解法利用微波辐射引起物体内部分子相互摩擦而产生热能,促使分子发生极化旋转。该方法水解快,一般4 h可以完成。
为了解决传统的酸水解法耗时长、步骤繁琐的问题,本实验以酸水解法为对照,微波水解法和高压水解法进行了比较,比较结果见表3。微波水解法与传统的酸水解法氨基酸测定结果之间无显著性差异,高压水解法测定结果中有个别氨基酸(精氨酸、组氨酸、甲硫氨酸、赖氨酸、缬氨酸、脯氨酸)的测定结果与传统的酸水解,微波水解法有显著性差异,大部分氨基酸无显著性差异。说明,微波水解法可以把水解时间从传统酸水解24 h缩短为4 h,并且检测步骤简单,易操作。

表3 三种不同水解方法比较结果Table 3 Three different hydrolysis methods comparison results
2.4.3 样品称样量的确定
为了确定不同的称样量对微波水解效果的影响,本实验分别称取猪肉0.1、0.2、0.5、0.6、1.0 g进行微波消解后测定17种氨基酸含量,结果见图3。称样量为0.5 g时水解效果最优,因此采用称样量为0.5 g。

图3 称样量对水解效果的影响Fig.3 Influence of sample weight on the hydrolysis effect
2.5 检出限
配制混合标准溶液质量浓度为5.0~375.0 ng/mL的标准溶液,测定峰面积,以质量浓度为横坐标、峰面积为纵坐标,绘制标准溶液的工作曲线,在此质量浓度范围内,标准品质量浓度与峰面积值均有良好的线性关系,检出限(limit of detection,LOD)以1/3信噪比评估法计算得出。具体线性方程及相关系数,检出限见表4。
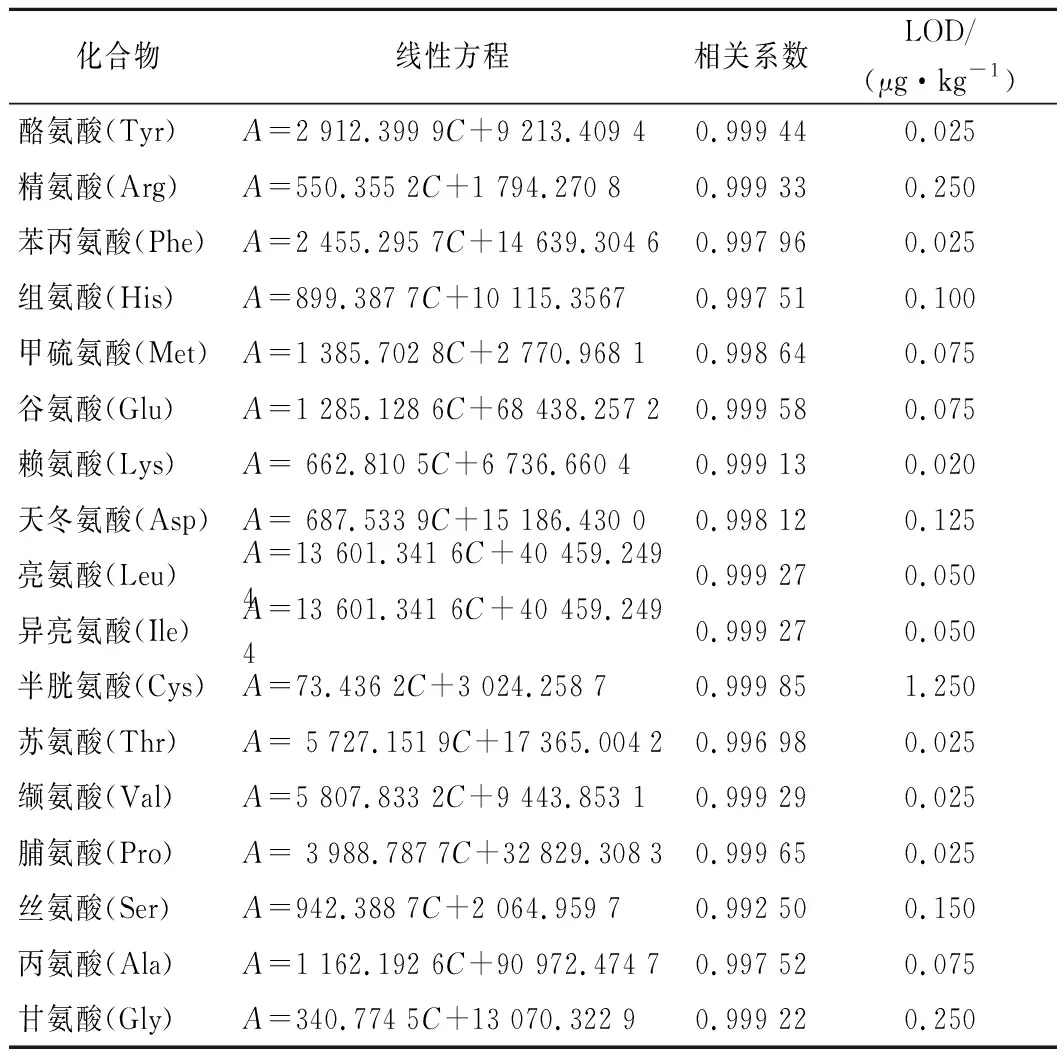
表4 十七种氨基酸的线性方程、相关系数(R)和检出限Table 4 The linear equation, correlation coefficient (R) and detection limit of 17 amino acids
2.6 准确度及精密度
采用添加回收的方法,测定不同动物源性食品基质中回收率,按表5中添加高、中、低水平做7次平行样品,测定精密度,结果见表5。不同动物源性食品基质中各氨基酸的回收率为85%~96%,精密度(relative standard deviation,RSD)为0.01%~0.71%。


续表5
2.7 LC-MS/MS与传统氨基酸分析仪方法比较
为了进一步验证LC-MS/MS测定动物源性食品畜禽肉和鸡蛋中氨基酸含量方法的可行性,与传统的氨基酸分析仪方法进行了比较,结果见表6。不同的动物源性样品采用氨基酸分析仪和LC-MS/MS测定结果无显著性差异,说明微波水解-LC-MS/MS法可以用于动物源性食品畜禽肉和鸡蛋中氨基酸含量的测定。

3 结论
本文采用微波消解技术消解畜禽肉和鸡蛋中蛋白质为游离氨基酸,以0.1%甲酸水溶液+10 mmol/L乙酸铵溶液和甲醇为流动相,梯度洗脱,串联质谱正离子模式进行监测,外标法定量,测定动物源性食品畜禽肉和鸡蛋中17种氨基酸。数据表明,本方法耗时短、操作较为简便、快捷、精密度高,样品分离度好,测定结果准确可靠,灵敏度高,所用试剂成本低,能满足动物源性食品畜禽肉和鸡蛋中17种氨基酸的检测。
猜你喜欢
中国中医急症(2019年10期)2019-05-21 07:20:46
天然产物研究与开发(2018年7期)2018-08-21 02:04:12
中国蜂业(2018年4期)2018-05-09 06:25:08
中学化学(2016年2期)2016-05-31 05:27:22
课程教育研究·下(2016年2期)2016-03-25 13:45:48
当代化工研究(2016年6期)2016-03-20 16:21:46
川北医学院学报(2015年5期)2015-12-05 08:22:43
中国医疗美容(2015年1期)2015-07-12 10:06:55
中医研究(2014年8期)2014-03-11 20:29:17
无机化学学报(2014年3期)2014-02-28 17:30:58
