
基于转录组学数据的抗真菌药物预测方法研究*
2023-02-20 02:48杨浩艺姚泽欢谭郁松
计算机工程与科学 2023年2期
杨浩艺,陈 微,姚泽欢,谭郁松,李 非
(1.国防科技大学计算机学院,湖南 长沙 410073;2.中国科学院计算机网络信息中心,北京 100190)
1 引言
以侵袭性真菌感染为代表的感染性疾病是导致发病率高和死亡率高的重要原因,尤其对于艾滋病毒感染患者以及患有其他共病的免疫力低下的患者造成了重大威胁。这些真菌造成的死亡率与耐药结核分岐杆菌造成的死亡率相当,超过了疟疾[1]。当前已知的对人类有致病性的真菌多达300多种,根据抗真菌药物的作用机制[2],已用于临床的抗真菌药物大致可分为多烯类、三唑类、烯丙胺类、棘白菌素和其他抗真菌药物。然而人类真菌感染的病理生理学研究仍然远落后于其他病原体引起的疾病[3]。此外,耐药菌的出现和广泛分布使得曾经容易治愈的疾病变得再次致命[4],以念珠菌为例,其对许多国家选择的标准抗真菌药物氟康唑以及新推出的棘孢菌素均具有耐药性[5]。因此,安全有效的抗真菌药物的研发显得十分必要且迫切。
本文利用CMAP(Connectivity MAP)[6]和LINCS(Library of Integrated Network-based Cellular Signatures)[7]高通量转录组学数据,基于WTCS(WeighTed Connectivity Score)算法[8],从已有抗真菌药物出发,发现其他药物潜在的抗真菌用途。本文通过将生物大数据应用于快速药物设计发现,基于生物医药数据特征构建端到端的药物预测策略,为加快新的抗真菌药物的研发进程提供计算方法。
2 数据驱动的药物重定位方法
根据Eroom定律[9],新药研发成本持续增长。面对从无到有的漫长传统药物研发过程,迅速积累的生物医学高通量数据推动了以生物信息学医药大数据为基础的系统性药物重定位[10]的发展,并引起研究人员广泛关注。药物重定位[11]针对已知的药物识别和发现其新用途、新疗效,是快速发现潜在药物的不错选择,既能够高效地找到目标药物,也可提前预知药物的副作用及用药注意事项。以抗真菌药物为例,在现有的、合成的或半合成的化合物库中进行抗真菌活性筛选,能够提前确定这些筛选出的化合物的最低毒性[1]。
数据驱动的药物发现方法依赖高质量的数据资源,GenBank数据库[12]整合了来自所有可用公共来源的DNA序列;PharmGKB数据库[13]为药物研发提供了潜在的药物-基因组关联信息以及基因型-表型信息;CMAP和LINCS数据库[6]提供了在不同细胞系中加入多种化合物所产生的基因表达谱;Drug Bank数据库[14]作为一个独特的生物信息学和化学信息学资源,结合了详细的药物/化学数据以及药物靶标/蛋白质信息;蛋白质结构数据库PDB(Protein Data Bank)[15]提供了目前最完整的蛋白质三维结构数据。
无论是传统的药物发现流程还是药物重定位,其关键点在于确定化合物的作用模式MOA(Mode Of Action)及其非靶点效应[16]。基于细胞转录反应检测药物作用模式(MOA)进而发现药物的方法所需信息量最少,且可以快速应用于新的化合物[11]。转录组数据能够直观反映在某一特定条件下基因表达、基因过表达或者基因沉默的情况,不同条件下转录组数据结果不尽相同[17]。针对药物发现来说,一是可以通过比较各种化合物作用于细胞与正常条件下细胞的转录组数据差异,找到有效药物;二是可以通过比较不同化合物在相同条件下作用于细胞的转录组数据,找到具有相同作用模式的化合物,进而达到发现潜在治疗药物的目的。在此背景下,利用基因表达谱、转录组谱以及生成的关联网络进行相关比对分析的方法以其快速高效、低成本的优势在临床治疗、药物作用模式阐述、药物重定位和系统生物学等多个研究领域得到了应用和发展[12]。
Lamb等[6]创建了大型的药物和基因标签公共数据库CMAP和LINCS,使得通过多种模式匹配算法挖掘生物数据特征之间的关联性成为可能[18]。他们采用L1000技术[19]基于大规模的统计分析辨识出人类细胞中978个基因作为全基因组的标志基因,进一步通过计算预测获得全部基因的表达量[20],实现了低成本、高通量的实验数据获取。到目前为止,LINCS计划已获得了77种典型细胞中的4 000多个沉默基因和7 000余种化学小分子刺激下的130余万个全基因组表达谱[21],为构建不同药物反应之间的关联关系奠定了牢固的数据基础。
3 抗真菌药物预测发现
本文针对抗真菌药物进行药物预测发现,以5种抗真菌分子化合物(氟胞嘧啶(flucytosine)、酮康唑(ketoconazole)、咪康唑(miconazole)、两性霉素B(amphotericin B)和制霉菌素(nystatin))为基础,基于CMAP数据库对5种药物与基因表达谱的联通性分数排序,针对每种药物确定一组上调和下调基因,每组包括10个基因。运用WTCS算法将查询药物的上、下调基因集与CMAP参考数据库中的扰动分子进行富集分析,计算相似性分数并对其进行排序,得到每种目标药物的相似药物列表。
考虑到选取的是不同种药物作用机制下的代表性药物,作用机制不同使得基因差异表达情况不同,综合分析5种抗真菌的药物特点及其相似药物列表结果之后,本文选择合并多个相似药物列表,利用RankAggreg[22]中的交叉熵-蒙特卡洛算法,分别采用Spearman footrule distance和Kendall’s tau distance 2种距离函数对5种相似药物列表进行聚合,对比2种距离函数得到的聚合结果筛选出抗真菌药物的预测药物列表。
3.1 DPA药物预测分析
基于Subramanian等[19]提出的WTCS计算相似性分数方法,本文提出药物预测分析DPA(Drug Prediction Analysis)方法,如图1所示。首先,通过CMAP数据库找到目标药物所对应的上、下调基因集,经过尝试与计算,将每组上、下调基因集中的基因标签数量控制在10个;其次,通过计算目标药物与CMAP参考标签数据库中每一个分子化合物扰动的相似性分数和差异显著性值,将得到的药物列表按照相似性分数由高到低进行排序,从而得到目标药物的相似药物列表;最后,采用RankAggreg中的2种距离函数方法对5种抗真菌药物的相似药物列表进行聚合,得到最可能的抗真菌药物预测结果。为了验证该方法的正确性,本文利用Glaser等[23]公布的HDAC(Histone Deacetylase)抑制剂的基因表达标签,对所设计的药物预测方法进行正确性验证。
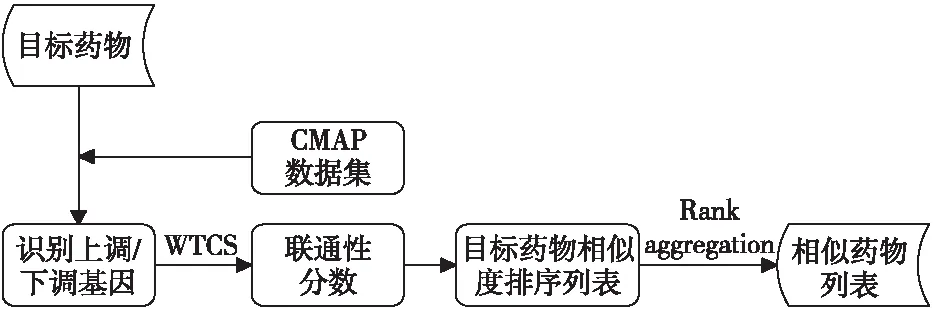
Figure 1 DPA drug discovery analysis process图1 DPA药物发现分析流程
3.2 WTCS相似性计算
经典的基因集富集分析GSEA(Gene Set Enrichment Analysis)[21]方法以查询目标化合物分子的基因标签作为输入,可以评估其与数据集中每个参考表达谱的相似性。给定所需要查询计算的目标化合物分子的基因标签(上调基因、下调基因),将目标化合物分子的基因标签与CMAP数据库中的编目列表进行比较分析,根据上调基因、下调基因在排序列表中的分布情况,可以将目标化合物的基因标签与数据库中的基因标签的关系分为正相关、负相关和无关3种。而正、负相关又可细分为强正(负)相关和弱正(负)相关。比对后可以得到目标化合物分子基因标签与数据库中化合物分子基因标签的联通性分数(Connectivity Score),相似性分数的取值在-1~1。
WTCS算法对传统的GSEA富集分析方法进行了改进,通过计算不同化合物分子基因标签的富集分数得到不同化合物的联通性分数,并通过联通性分数的高低找到与目标药物作用相似的药物,进而达到重定位药物的目的。该算法原理相对简单,易于操作实现,在寻找相似化合物分子计算中已有广泛的应用。
WTCS算法基于Kolmogorov-Smirnov富集统计ES(Enrichment Score)的非参数性相似性度量,对于输入基因集(qup,qdown),按照式(1)的计算方式得到与某一参考基因标签的相似性分数Wq,r:
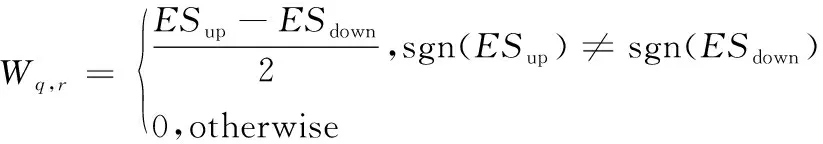
(1)
其中,ESup、ESdown分别是qup、qdown在参考基因标签下的富集分数。
3.3 Rank aggregation加权聚合排序
Rank aggregation是一种对多个排序列表进行整合得到一个综合排序列表的算法[22]。在该算法中,Spearman footrule distance距离函数根据不同排序列表内元素的排序位置进行距离计算,该方法简单且所需信息量少;而Kendall’s tau distance距离函数需要联合不同排序列表中对应的元素对距离进行综合计算,该方法复杂但最终得到的结果列表排序等级差异明显。Rank aggregation算法在R语言中有现成的包RankAggreg可用,因此实验过程中只需要直接输入需要聚合的排序列表,并调整迭代次数、距离函数等参数信息,使得结果收敛到最小。
4 结果
4.1 数据和方法
首先利用R语言下载CMAP数据集,该数据集中包含多种化合物作用下不同细胞系细胞的基因表达谱。这些化合物可以是蛋白、小分子化合物或者是复杂化合物。通过查询氟胞嘧啶、酮康唑、咪康唑、两性霉素 B和制霉菌素的ID号,可以确定在这些化合物作用下细胞的基因表达情况,根据不同基因表达结果的排序筛选得到5种化合物的基因标签集。本文的药物相似性计算在整个CMAP数据集上针对所有化合物进行相似性计算分析。GSEA和WTCS算法的计算流程由Bioconductor[24]中的piano包(使用各种统计方法从不同的基因统计水平和广泛的基因集合进行基因集分析)实现,通过分别对上调基因和下调基因进行富集分析,可以得到所要查询的基因标签与参考数据库中的基因标签的联通性分数,以及差异显著性值(P值)。药物发现分析流程利用基于R语言实现的Bioconductor开源环境下的PharmacoGx[25]、piano、bioMaRt[26]和RankAggreg[22]等R语言包实现。
4.2 药物相似性分析结果
在针对5种抗真菌药物分别计算得到的排名前10的相似药物列表结果(表1)中,酮康唑和咪康唑的相似药物列表结果相似程度较高,酮康唑相似药物列表中相似性分数最高的可达0.82,而咪康唑相似药物列表中相似性分数最高可达0.88,这2种药物的相似药物列表在后续的实验中值得关注;与酮康唑和咪康唑的相似药物列表相比,其余3种药物的相似药物结果相似程度相对较低,但前10位最相似药物的相似性分数均在0.6以上。从实验结果的准确性上看,上述药物计算结果中,酮康唑和咪康唑的相似药物结果不仅在相似性分数上表现较好,其结果的P值也几乎全在0.01以内;而两性霉素B、制霉菌素和氟胞嘧啶的计算结果虽然在相似性分数和差异显著性值上表现相对较差,大部分计算结果的P值均大于0.01,但除去极个别药物外,这些相似药物结果的P值均小于0.05,仍然具有一定的参考价值。5种目标药物计算得到的P值结果如表2所示。从这些相似药物结果也可以看出,由于不同的药物作用机制不同,不同药物的相似药物以及其相似程度也存在明显差异。
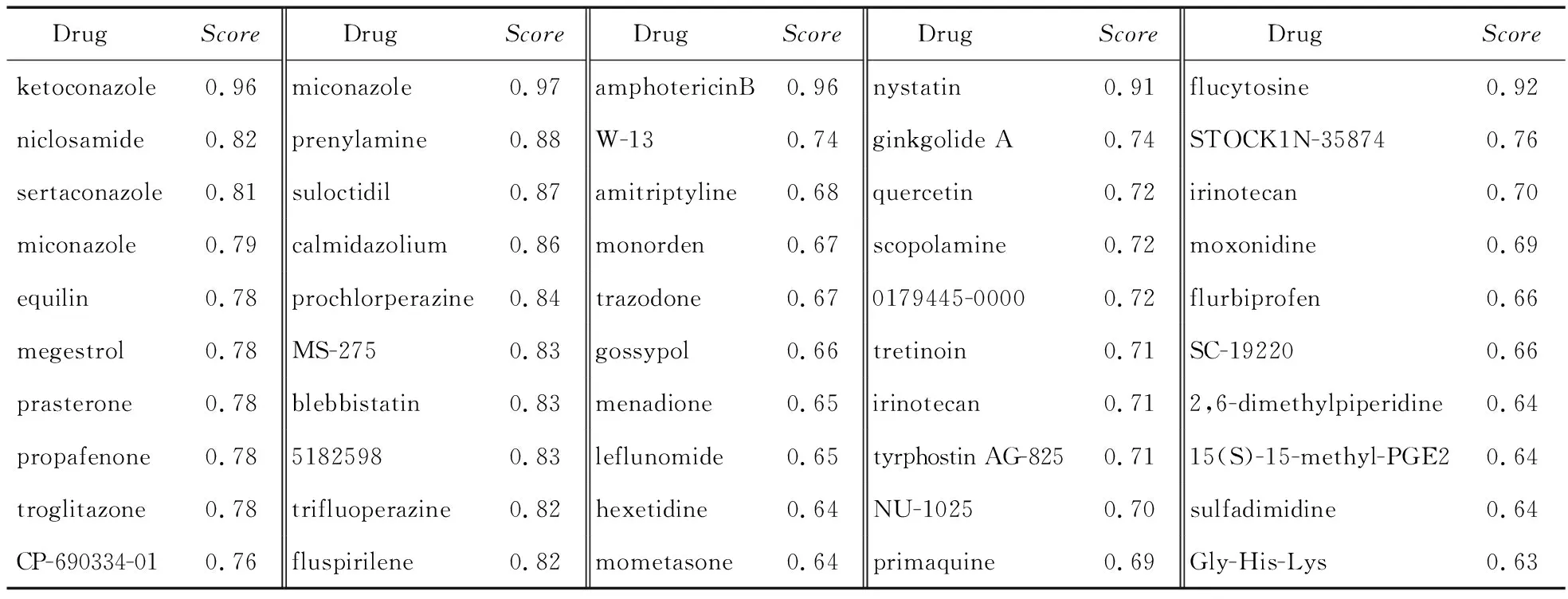
Table 1 List of inferred antifungal candidates表1 预测抗真菌候选药物列表
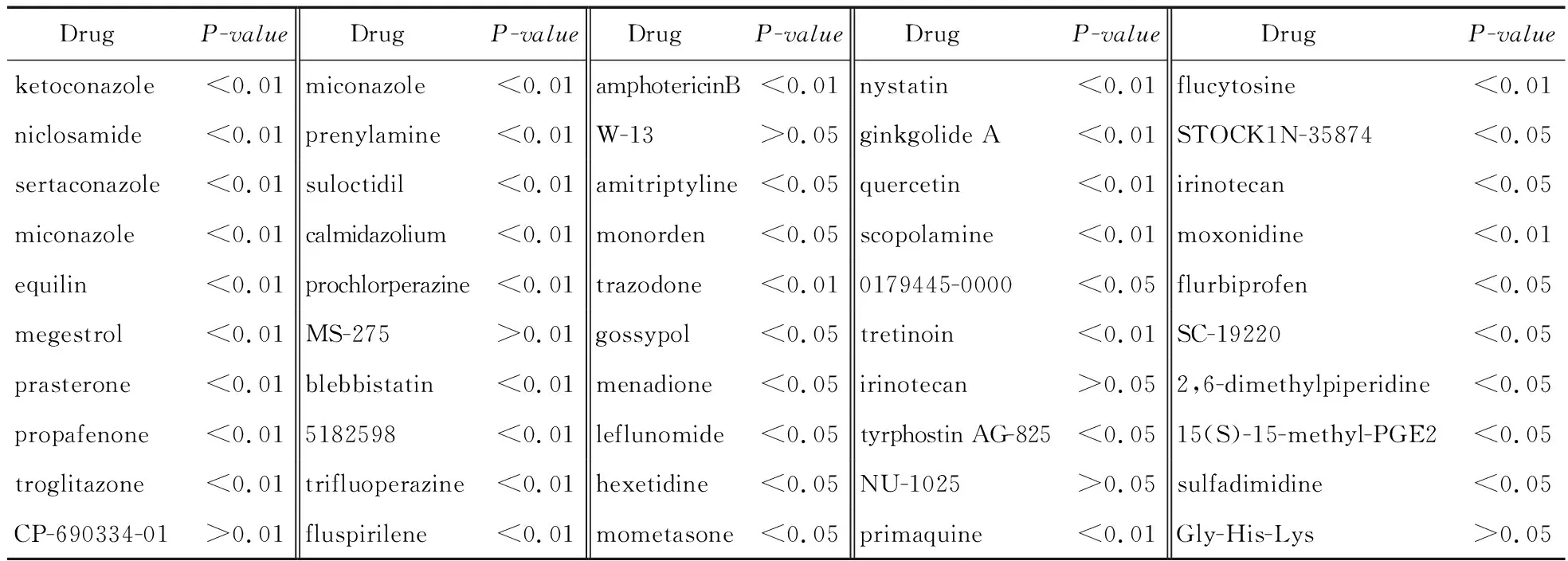
Table 2 P-value list of inferred antifungal candidates表2 预测抗真菌候选药物P值列表
4.3 抗真菌药物预测结果
在5种药物的相似药物结果的基础上,本文利用RankAggreg中的交叉熵-蒙特卡洛算法,分别采用Spearman footrule distance和Kendall’s tau distance 2种距离函数对5种药物的相似药物列表结果进行聚合排序。在4.2节得到的相似药物排序结果中,根据化合物分子与目标药物的正负相关性最终得到的相似性分数存在0~1,0,-1~0这3种情况。由于本实验中只考虑正相关的情况,对于每种药物的相似药物列表只选择相似性分数大于或等于0的化合物分子进行聚合排序,在实验过程中不断调整迭代次数、分位数和生成样本数量等参数以获得更加精确的收敛结果。由于计算方法的差异,采用Spearman footrule distance距离函数和Kendall’s tau distance距离函数最终收敛得到的结果数值水平存在一定差异(如图2所示),但利用2种距离函数聚合得到的药物列表排序结果相似。通过排除掉5种目标查询药物,最终选择了10种化合物分子作为抗真菌药物预测药物结果(如表3所示),分别是伊利替康(irinotecan)、氯硝柳胺(niclosamide)、舍他康唑(sertaconazole)、普尼拉明(prenylamine)、银杏内酯A(ginkgolide A)、莫索尼啶(moxonidine)、槲皮素(quercetin)、舒洛地尔(suloctidil)、卡米达佐(calmidazolium)和STOCK1N-35874(详细计算分析结果参见https://github.com/yeaouh/antifungal)。

Table 3 Prediction results of antifungal candidates 表3 抗真菌候选药物预测结果

Figure 2 Convergence results of RankAggreg图2 RankAggreg收敛结果
5 结束语
本文基于WTCS、GSEA算法以及所设计的药物发现分析方法对5种具有代表性的抗真菌药物进行了相似药物预测,并通过综合排序得到了抗真菌药物最相似药物列表,最高相似度为88%,得到的抗真菌相似药物列表具有一定的参考价值。在最终计算得到的抗真菌药物预测结果中,经过文献查证,已经证明舍他康唑[27]、舒洛地尔[28]能够应用于临床抗真菌药物治疗。另外,根据文献显示,一些抗氧化(如槲皮素)、抗癌(如伊利替康、槲皮素)等药物的作用机理可能同样适用于真菌[1],从而达到治疗真菌感染的目的,但这些猜测显然需要实验研究进行确认。
利用高通量组学数据对现有药物进行筛选分析能够大大缩短药物发现流程。通过计算得到的理论结果有待进一步的细胞、动物及临床实验验证。下一步将基于更大规模的数据集进行探索,寻找使得预测结果更加可靠的方法,以及探索深度学习技术在组学数据分析处理、药物设计发现中的应用,实现更为精准的抗真菌药物预测发现。
猜你喜欢
数学物理学报(2022年5期)2022-10-09
中学生数理化·七年级数学人教版(2022年11期)2022-02-14
小学生学习指导(中年级)(2021年4期)2021-04-27
河北画报(2020年8期)2020-10-27
课堂内外(初中版)(2020年5期)2020-06-19
科普童话·学霸日记(2020年1期)2020-05-08
小天使·一年级语数英综合(2019年2期)2019-01-10
浙江大学学报(工学版)(2016年2期)2016-06-05
中学生数理化·中考版(2015年10期)2015-09-10
华东师范大学学报(自然科学版)(2014年3期)2014-03-11
