
Electric field-driven folding of single molecules
2023-02-18 01:55SaisaiYuanYuZhouTengyangGaoLichuanChenWeiXuPingDuanJuejunWangZhichaoPanChunTangYangYangRuiyunHuangZongyuanXiaoWenjingHong
Chinese Chemical Letters 2023年12期
Saisai Yuan ,Yu Zhou ,Tengyang Gao ,Lichuan Chen ,Wei Xu,Ping Duan,Juejun Wang,Zhichao Pan,Chun Tang,Yang Yang,Ruiyun Huang ,Zongyuan Xiao ,Wenjing Hong
State Key Laboratory of Physical Chemistry of Solid Surfaces,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,China
Keywords:Electric field Single-molecule Break junction Molecular folding In-situ switch
ABSTRACT Folding of molecules is an essential process in nature,and various molecular machines achieve their chemical and mechanical function via controlled folding of molecular conformations.The electric field offers a unique strategy to drive the folding of molecular conformation and to control charge transport through single molecules but remains unexplored.The single-molecule break junction technique provides access to detect the conformational changes via the monitoring of single-molecule conductance,and the electric field between two metal electrodes with nanoscale spacing can provide an extremely strong to achieve in-situ control and detection of molecular folding at the single-molecule level.Here,we use the electric field to control the single-molecule folding using the scanning tunneling microscope break junction (STM-BJ) technique.The electric fields induced folding could lead to a ∼1400% conductance change of the single-molecule junctions,and the folding/unfolding process can be in-situ switched at the scale of milliseconds.DFT calculations suggest the conformational control originates from the electric fieldinduced charge injection,and the formation of homoconjugated conformation with the overlapped orbitals.This work provides the first demonstration of electric field-driven molecular folding,which is essential for the understanding of molecular machines in nature and for the design of artificial molecular machines.
The different conformations of the molecules lead to the variation of their electrical features,and controlling the conformations of the molecules provides the electric field-induced folding functions in molecular machines [1].The optical,chemical,and mechanical approaches are added to generate metal coordination bonds [2],hydrogen bonds [3] and photo-induced isomerization[4] to change the molecular conformational [5–9],which could be characterized with ensemble methods such as NMR and UV–vis spectra [8,9].Thein-situcontrol of molecular folding in nature is mostly achieved by electrical stimulation [7,10,11].However,the electric field control of molecular folding remains unexplored,which is mainly due to the experimental challenges in the investigation of electric-field responsive molecular systems at the molecular level.The single-molecule break junction technique provides access to detect the conformational changesviathe monitoring of single-molecule conductance,and the electric field between the two metal electrodes with nanoscale spacing can provide a field intensity of up to 108–109V/m [12],providing a promising strategy to achievein-situcontrol and detection of molecular folding at the single-molecule level [13,14].
The external electric field has demonstrated the potential to accelerate chemical reaction and assembly at the single-molecule scale [15–19],and our recent study [20] demonstrates that intramolecular rotation induced by electric fields can enhance the stacking of phenyl rings [21].However,the slight changes in conformation are not enough to cause detectable conductance changes,leading to the challenge of using the electric field to control molecular conformation at the single-molecule level.To overcome this challenge,we designed a simple primitive molecule with homoconjugated conformation,which will significantly increase the conductance difference between the unfolding and folding states.The structure of the model molecule is shown in Fig.1a,and two phenyl rings linked by methylene are a primitive model in which the weak interaction varied by the rotation of phenyl rings through theσbonds.When the two benzene rings fold along theσbonds to face-to-face,the homoconjugation interaction,which represents the orbital overlap of twoπ-systems separated by a nonconjugated group such as CH2[22–26],will act as ‘folding’and leads to the pronounced conductance difference of the singlemolecule junctions (Fig.1b).
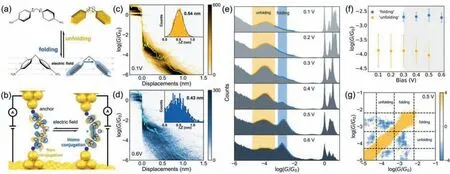
Fig.1. Single-molecule conductance measurements.(a) The molecule structures and the scheme of electric field-controlled molecular folding.(b) The STM-BJ setup and the switching process between the two states with different conformations.Measurements of single-molecule conductance.(c,d) Two-dimensional conductance-displacement histograms,and displacement distribution (inset) of 4,4'-diaminodiphenylmethane (DDM) with different bias voltages.The yellow and blue curves are the typical individual conductance traces and the detail information are shown in Fig.S3b.(e) One-dimensional conductance histograms of DDM with different bias voltages.(f) Conductance of the ‘unfolding’and ‘folding’states with different bias voltages.(g) Correlation analysis between the ‘unfolding’and ‘folding’states at 0.5 V.
Here,we demonstrate the electric field-driven molecular folding at the single-molecule level using the scanning tunneling microscope break junction (STM-BJ) technique (Fig.S1 in Supporting information) [27].We investigate the conductance of the singlemolecule junction with various bias voltages and find two distinct folding states with different bias voltages.We further reveal that the high conductance state shows the mechanism of throughspace charge transport,while the low conductance state shows the mechanism of through-bond charge transport.DFT calculations combined with a series of control experiments suggest that a strong electric field can induce the charge injection,which further leads to the formation of the homoconjugation transport.This work reveals the potential for investigating molecular machines using single-molecule electrical characterization,which is of interest to synthetic,physical and materials chemists working in relevant areas.
To determine the single-molecule conductance of different states,we measured the conductance of 4,4'-diaminodiphenylmethane (DDM) with a diphenylmethane (DPM)backbone and bilateral amino anchors using the STM-BJ technique [28–31] at room temperature.We dissolved DDM in 1,2,4-trichlorobenzene (TCB) and measured the conductance at various bias voltages applied between the gold tip and gold substrate.The data analysis was processed in our open-source code XME analysis (https://github.com/Pilab-XMU/XMe_DataAnalysis,XMe-data analysis,and the detailed process of data processing is provided in methods section in Supporting information).As shown in Fig.S3b (Supporting information),the individual conductance traces show conductance plateaus at around 10−4G0.Here,G0is the quantum conductance and is 77.6 μS under a 0.1 V bias voltage[32].Interestingly,a new conductance plateau appears around 10−3G0when the bias voltage is increased to 0.6 V.The results of higher bias voltage are shown in Fig.S4 (Supporting information).The two-dimensional conductance histograms in Figs.1c and d have shapes similar to those of the individual conductance traces in the corresponding ranges.To extract the conformations of the molecular junctions,the displacement distribution histograms at corresponding ranges were also analyzed.As shown in the insets in Figs.1c and d,after considering the snap-back distance of gold electrodes (0.5 nm) [33],the actual junction lengths for the low and high bias voltages are 1.04 (0.54+0.5) nm and 0.93(0.43+0.5) nm.These lengths are close to the fully extended lengths of the molecules (0.97 nm),indicating that gold electrodes coordinate with amino groups of the molecules with different bias voltages.There is also a difference of 0.1 nm in the characteristic length of molecular junctions under high and low bias voltages,suggesting the changes of the molecular conformations at high bias voltages,which is also confirmed by the DFT results (Fig.S14 in Supporting information).
The most probable conductance with different bias voltages varying from 0.1 V to 0.6 V were determined from the conductance histograms,as shown in Fig.1e (the corresponding twodimensional conductance histograms are shown in Fig.S5 in Supporting information).A sharp peak aroundG0arises from the formation of the Au-Au atomic contact [34],and the conductance peaks in the range of 10−5–10−3G0indicate the conductance signals from the single-molecule junctions at the 0.1 V bias voltage.A new distinct peak in the blue-ribbon area appears when the bias voltage reaches 0.4 V.With the increase of bias voltage,the ‘folding’state becomes increasingly prominent and begins to dominate at 0.6 V.The main peak center,determined by Gaussian fitting at 0.6 V,is located at 10−2.73G0(the ‘folding’state),which is ∼1400%higher than the conductance at 0.1 V (10−3.88G0,the ‘unfolding’state).We also investigated the change changes with the negative bias voltages (Fig.S6 in Supporting information),and the trend is consistent with that of positive bias voltage.
To further confirm the appearance of the ‘folding’state at high bias voltages,we analyzed the variations of conductance with different bias voltages.As shown in Fig.1f,the variation of the ‘folding’state is smaller than that of the ‘unfolding’state,indicating that the rotation in the ‘unfolding’state is restricted.To exclude the conductance switching from different anchoring positions of the same molecule,for example,the phenyl rings of DDM being absorbed,like 4,4'-bipyridine [35,36] on the surface of Au forming an Au-πbond,a correlation analysis [37,38] was performed (Fig.1g).There is a significant anti-correlation at the intersection of the region ‘off’and region ‘on’,indicating that the molecular junction is constructed either by a molecule at the ‘unfolding’state or one at the ‘folding’state,but not a molecule with different anchoring configurations.
We further performed the cycle characterization between 0.1 V and 0.6 V bias voltages to confirm this reversibility of the electric field-induced molecular folding.We collected traces to construct each of the one-dimensional and two-dimensional conductance histograms (Figs.S7 and S8 in Supporting information).The dominant conductance peaks were determined from the three subsequent switching cycles (Fig.2a),indicating that the singlemolecule conformations based on the DDM junction could be controlled reversibly with bias voltages.We further performed the bias-modulation experiment at room temperature to evaluate thein-situswitching characteristics.A hovering experiment was conducted by holding a molecular junction for 600 ms for bias modulation between 0.1 V and 1.0 V with a pulse length of 100 ms.More information is available in the methods section in Supporting information.In Fig.2b,a typical individual switching trace is shown as a black line and the conductance can be modulated by bias voltages.We selected approximately 200 traces to construct the twodimensional conductance histograms,and we found that the junctions can be cycled reliably between the ‘unfolding’state and the‘folding’state during the bias-modulation.The values of the low and high conductance are around 10−3.8G0and 10−2.8G0,which is consistent with theex-situresults.
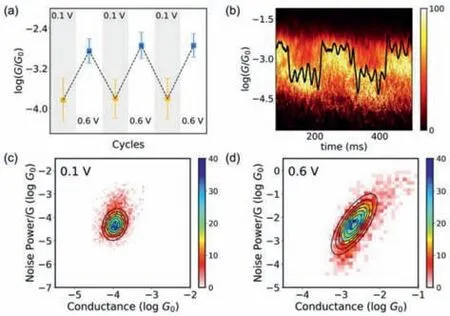
Fig.2. Switching performance of single-molecule junctions.The detailed measurement and data processing are described in the methods section in Supporting information.(a) Plots of the dominant conductance with three subsequent switching cycles.The errors are the standard deviation of the Gaussian fitting from corresponding one-dimensional conductance histograms.(b) Two-dimensional conductance histograms for the in-situ and reversible switching performance.The black curve is a typical individual switching trace.Two-dimensional histograms of normalized flicker noise power versus the average conductance of DDM with different bias voltages,(c) ‘unfolding’state,(d) ‘folding’state.The noise power scales are determined to be G1.4 for the ‘unfolding’state and G2.0 for the ‘folding’state.
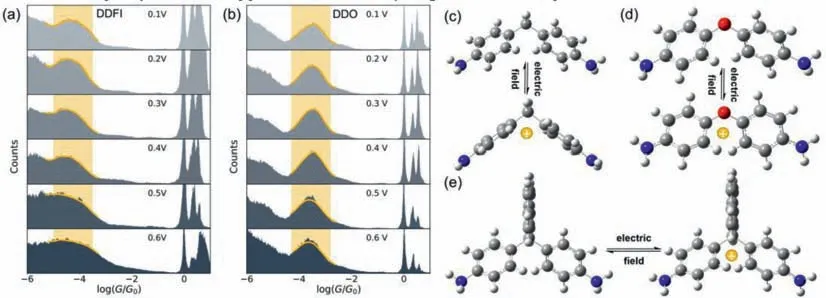
Fig.3. One-dimensional conductance histograms under different bias voltages.(a) DDFI,(b) DDO.The conformations of the ground states and the charge injected states: (c)DDM,(d) DDO,(e) DDFI.
To further identify the molecular conformation,we performed power spectral density (PSD) analysis [39,40] for the singlemolecule junctions at the ‘folding’state and the ‘unfolding’state.To obtain the flicker noise signal,we paused the piezo once the molecules had been caught and then analyzed the noise signals using a fast Fourier transform (FFT) algorithm.According to the principle of PSD analysis [41],the correlation between noise power and average conductance can be used in a qualitative determination of the transport mechanism of molecular junction (details are provided in the methods section in Supporting information)[42].As shown in Figs.2c and d,the noise power of ‘unfolding’state scales isG1.4,whereGis the mean conductance,indicating that the charge transport tends to the through-bond mechanism[41].On the contrary,the noise power of the ‘folding’state scales asG2.0,indicating that the through-space mechanism dominates the charge transport.The change from through-bond transport to through-space transport indicates the presence of different conformations in both states,suggesting a new through-space pathway introduced by the interaction of the phenyl rings [26].
DDO (4,4'-oxydianiline) and DDFI (4,4'-(9-fluorenylidene)dianiline) are used as control molecules to reveal the mechanism of the molecular folding (Figs.S15b and c in Supporting information).We carried out the single-molecule measurement of DDO whose phenyl rings can fold freely.As shown in Figs.3a and b,the conductance remains unchanged from 0.1 V to 0.6 V,indicating that the electric field does not change the conformation directly.We also performed the simulation results of DDM with various electric fields.As shown in Fig.S16 (Supporting information),only when the direction of the electric field is y-axis,the homoconjugated conformation will form.However,the dominant direction of the electric field isx-axis in the molecular junctions,and there are insufficient electric fields along they-axis to change the conformation.Inspired by previous works [43,44]that the electric field can induce the charge injection behavior,we calculate the charged conformation of DDM,DDO and DDFI.As shown in Figs.3c-e,the conformation of the positive DDM changes from the twist state to gable state,while the conformation of the positive DDFI remains the twist state.For DDFI molecule,the high steric hindrance of the fluorene group inhibits the rotation of two phenyl rings.Although the phenyl rings of DDO are free,the positive DDO remains twisted,which is consistent with the experimental result.As shown in Fig.S17 (Supporting information),p-πconjugation formed from oxygen and phenyl rings,leading to the coplanar tendency of two benzene rings.Based on the above reasons,only DDM molecules show the folding phenomenon.Based on the above analysis,the electric field induced charge injection is suggested to be the key step for the folding.
To further confirm that the folding comes from the electric field effect,the single-molecule conductance measurement was performed in propylene carbonate solvent.As shown in Figs.S9 and S13 (Supporting information),the high conductance does not appear until the bias voltage approaches 0.5 V.This indicates that the polar solvent weakens the strength of the electric field on the molecule,inhibiting the generation of the high conductance state.In addition,the high conductance still appeared at −0.6 V,suggesting that the redox process did not occur in the experiment (Fig.S10 in Supporting information).Thus,the electric field plays an important role in conductance switching.To exclude the possibility that reaction occurs only under a strong electric field[12,45,46],we also examined a deprotonation mechanism controlling the conformation (Figs.S18 and S19 in Supporting information).It was found that the kinetic barrier is indeed allowed at room temperature whether water is involved or not,and the introduction of an electric field makes the thermodynamics of the reaction more favorable (Fig.S20 in Supporting information).Indeed,the amino groups are the only Lewis base in the system to capture the hydrogen proton.To confirm this hypothesis,we synthesized a reference molecule with methylthio anchoring groups(DMMeS) and measured the single-molecule conductance.The result (Figs.S11 and S15d in Supporting information),indicates that the high conductance still is observed even in the absence of the amino groups,which allows rejection of the hypothesis.In addition,the enhancement of conductance of the control molecule DMMeS at higher bias voltage eliminates the possibility of increasing conductance from Au-N formation [47] in the strong electric field.
Combining DFT and the non-equilibrium Green’s function method (NEGF) integrated within the Quantum Atomistix ToolKit(ATK) software package [48],we investigated the mechanism of the conformation changes under electric field.First,we optimized the geometries of the devices and then estimated their transmissions.Details of these calculations are provided in the methods section in Supporting information.We performed the frontier molecular orbitals of the ground (‘unfolding’) state and the charged (‘folding’) state (Fig.4a).The ‘folding’state generates the overlapped orbitals and leads to a new electronic transition [24,26].This result indicates the more delocalized electron distribution and stronger transmission of the gable conformation.In addition,the injected state provides more active charge for the transport.Therefore,this work shows the particularly high conductance in throughspace transport.As shown in Fig.4b,we calculate the transmission curves of the ground state and the charge-injection state under zero bias.For the positive charge injection,the Fermi energy should be close to the HOMO and the conductance of the homoconjugated state should be higher than that of the nonconjugated state.Therefore,the Fermi level is probably around the green ribbon regime (Fig.4b).To determine the relative position of Fermi energy with respect to the HOMO energy,thermoelectric voltages are measuredviahome-built STM-BJ [49,50],and the details of the measurement are shown in the methods section in Supporting information and histograms of thermoelectric voltage measurements for four temperatures are shown in Fig.S12 (Supporting information).Previous studies suggested that the positive value of the Seebeck coefficient indicates the Fermi energy is close to the HOMO,and a negative value means the Fermi energy is close to LUMO [51,52].As shown in Fig.4c,we measured the thermopower by characterizing the thermoelectric voltages generated from four temperature differences between the tip and the substrate.The Seebeck coefficient of our molecule is determined to be ∼33.4 μV/K,indicating that the Fermi energy position is close to the HOMO.Together with the experimental conductance difference,the Fermi energy is suggested to be in the range labeled by the green ribbon regime (Fig.4b) that the barrier of charge injection is less than ∼1 eV.
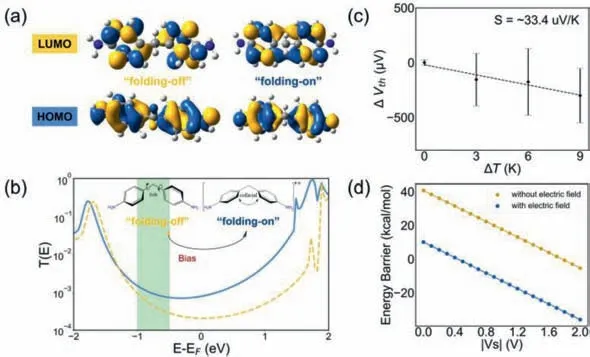
Fig.4. Theoretical results for the charge transport.(a) Frontier molecular orbitals of two conformations,and the isovalue is 0.02.(b) The transmission curves at zero bias voltage.The green ribbon regime shows the possible range of the Fermi energy.The homoconjugated conformation is injected with a single+charge,and the nonconjugated state is in the ground state conformation.(c) The Seebeck coefficients obtained from the thermoelectric voltage as a function of ΔT=+0 K,+3 K,+6 K and +9 K.The dashed line is the linear fitting lines.The error bars are based on the standard deviation in the Gaussian fitting of thermoelectric voltages.(d) The calculated energy barrier.Blue and yellow curves represent the cases with and without the oriented external electric fields (OEEFs) along with two nitrogen atoms (F=0.005 a.u.),respectively.
To evaluate the energy barrier quantitatively,we calculated the relevant redox barrier and proposed a reliable charge injection mechanism.From the energy point of view,the oxidation reaction process is Initial →OX+e−,and the oxidation barrier isΔE(OX)=E(OX)+E(e−) -E(Initial).While the electron energy isE(e−)=μ+eVs,whereμis the chemical potential of the gold electrode without bias (about −5.3 eV),and e is the electron charge(-1.6×10−19C),Vsis the base electrode potential.As shown in Fig.4d,the oxidation barrier was calculated to beΔE(OX)=40.72–23.02|Vs| (kcal/mol) and plotted as the yellow curve without electric field by DFT.This means that the oxidation reaction occurs spontaneously only when the bias exceeds 1.77 V,which is much higher than the bias applied in the experiments.To further understand the difference,there is a very strong electric field in the space of the electrode [12] that we further calculated the oxidation barrier under the electric field.As shown in Fig.4d,with the electric field of 0.005 a.u.(i.e.,2.57 V/nm) [53,54],the oxidation barrier is calculated to be 10.13–23.02|Vs| (kcal/mol) and plotted as the blue curve,which is much lower than the barrier without the electric field,and indicates that the charge injection occurs spontaneously when the bias exceeds 0.44 V.We speculate the reason why the electric field can reduce the oxidation barrier is that the electric field can stabilize the charged oxidation state far beyond the initial neutral state.Thus,with the increase of the bias,the proportion of the oxidation state gradually increases.When the bias reaches 0.6 V,the oxidation barrier is −3.69 kcal/mol under the electric field of 0.005 a.u.,and the oxidation state in the system would be utterly dominant according to the Boltzmann distribution,which is consistent with the experiments.Recently Stoddartetal.have also demonstrated that the electric field can induce the charge injection and promotes the dehydrogenation reaction at room temperature [43,44],which further suggests the charge injection could occur under the electric field.The thermoelectric experiment and theoretical calculation suggest that the charge injection could occur in our experimental condition,and the applied electric field further reduced the oxidation barrier.
To further confirm that the high conductance comes from the homoconjugated conformation,we compared the transmission curves of gable DDM and 7,7'-diphenylnorbornane with amino groups (DAPN,Fig.S21 in Supporting information).As shown in Fig.S15a (Supporting information),the transmission curve of the homoconjugated state is similar to that of DAPN with almost the same conductance at the Fermi energy level,owing to the similar conformation (the gable conformation) of the ‘folding’state and DAPN.We speculate that the strong electric field can make the phenyl ring folding to form the homoconjugated conformation and change the conductance.So,we performed the singlemolecule measurement of DDFI whose conformation is locked by its highly sterically hindered structure.As shown in Fig.3a,the conductance remains unchanged at high bias in the conductance measurement.These results demonstrate that the if the phenyl rings on both sides of methylene can fold freely,and the electric field can change their conductance.
To investigate the thermodynamic differences between the twist (‘unfolding’state) and gable conformations (‘folding’state),we calculated their single point energies of them at the neutral state and positive state,respectively.When both are neutral,the twist conformation is more stable than the gable conformation by 6.75 kcal/mol,which indicates that the twist conformation is absolutely dominant over the gable conformation according to the Boltzmann distribution.When both are positive,the gable conformation is more stable than the twist conformation by 8.93 kcal/mol,which indicates that the gable conformation is absolutely dominant over the twist conformation according to the Boltzmann distribution.Some earlier publications have also pointed out that with a strong electric field,the charge would be injected into the molecules [43] and the charge-injected state,or cation can be stabilized by homoconjugation [55–57].This is also supported by the switching of charge transport mechanism,which is from through-bond at the ‘unfolding’state to through space at the ‘folding’state.Based on the above discussion,the molecular folding mechanism involves the electric field-induced charged state and conformational changes to the ‘folding’state with homoconjugation,which leads to an increase of single-molecule conductance and a decrease of conductance variation.
In conclusion,we demonstrated the electric field-controlled molecular folding and unfolding using the STM-BJ technique.We found that molecular folding/unfolding controlled by the strong electric field leads to the switch between distinct ‘unfolding’and‘folding’conformations in the single-molecule junctions.The electric field-induced homoconjugated conformation provides a new through-space pathway and leads to a ∼1400% enhancement of conductance.The DFT results indicate that the homoconjugated conformation is formedviaa strong electric field-induced charge injection that the applied electric field significantly reduced the energy barrier,and the transmission probabilities are consistent with experimental results.This work provides a new strategy for the control of molecular folding for the future design and fabrication of molecular machine and responsive molecular materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Nos.22250003 and 22205084),National Key R&D Program of China (No.2017YFA0204902).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108404.
 Chinese Chemical Letters2023年12期
Chinese Chemical Letters2023年12期
- Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers