
Iron/B2pin2 catalytic system enables the generation of alkyl radicals from inert alkyl C-O bonds for amine synthesis
2023-02-18 01:55YnqingZhuShuiChenZhenZhouYunHeZhengliLiuYngLiuZhngFeng
Chinese Chemical Letters 2023年12期
Ynqing Zhu ,Shui Chen ,Zhen Zhou ,Yun He ,Zhengli Liu,c,∗ ,Yng Liu ,Zhng Feng,c,∗
a Chongqing Key Laboratory of Natural Product Synthesis and Drug Research,School of Pharmaceutical Sciences,Chongqing University,Chongqing 401331,China
b Department of Medicinal Chemistry,School of Pharmacy,Fujian Medical University (FMU),Fuzhou 350005,China
c Affiliated Hospital of North Sichuan Medical College and Medical Imaging Key Laboratory of Sichuan Province,Nanchong 637503,China
Keywords:C-N bond formation Reductive amination Borane reagent Iron catalysis Green chemistry
ABSTRACT A method for the generation of alkyl radicals from inert alkyl C-O bonds has been developed via an iron/borane reagent/alkoxide catalytic system,which can be employed for the synthesis of amines from nitroarenes with excellent efficiency.This reductive amination features good functional group compatibility and enables the late-stage amination of bio-relevant compounds,thus providing good opportunities for applications in medicinal chemistry.Preliminary mechanistic studies reveal that the amine synthesis may be involving a Fe/Li cation-assisted single electron transfer pathway to form alkyl radicals,and the low-valent iron species may be the active intermediates.
Alkyl radicals have received increasing attention in transition metal-catalyzed transformations in recent years [1].At present,the generation of alkyl radicals is typically from alkyl halides,while a few alkyl radical sources have been reported,such as alkyl carboxylic acids [2,3] and alkylamines [4].However,these approaches suffer from some limitations,including halogenated wastes and the requirement of multi-step synthesis of radical precursors.Therefore,developing an efficient method for the generation of alkyl radicals from green feedstocks is highly desired.Alkanols are more popular starting materials than alkyl halides in organic synthesis because of their halogenated-waste avoidance,good safety and ready availability.Alkanols as radical sources in the Giese reactions and Ni-catalyzed coupling reactions have been reported,in which alkanols should be converted into redox-active radical precursors(Fig.1A) [5–13].In contrast,alkyl pseudohalides,such as alkyl tosylates are attractive alkyl group sources as they avoid multi-step pre-functionalization.Nevertheless,alkyl tosylates are inert and difficult to generate alkyl radicals through the SET process due to the high-lying antibonding orbital of the C-O bond [14].Very recently,Weix [15] and Komeyama [16–18] found that highly nucleophilic cobalt(I) species can undergo SN2 reaction with alkyl tosylates to afford cobalt(III)-alkyl complexes,which then provide alkyl radicals through the homolytic cleavage of Co-C bond (Fig.1B).In order to enhance the synthetic utility of alkanols,we set out to develop new catalytic systems for efficient generation of alkyl radicals from inert C-O bonds [19–21].
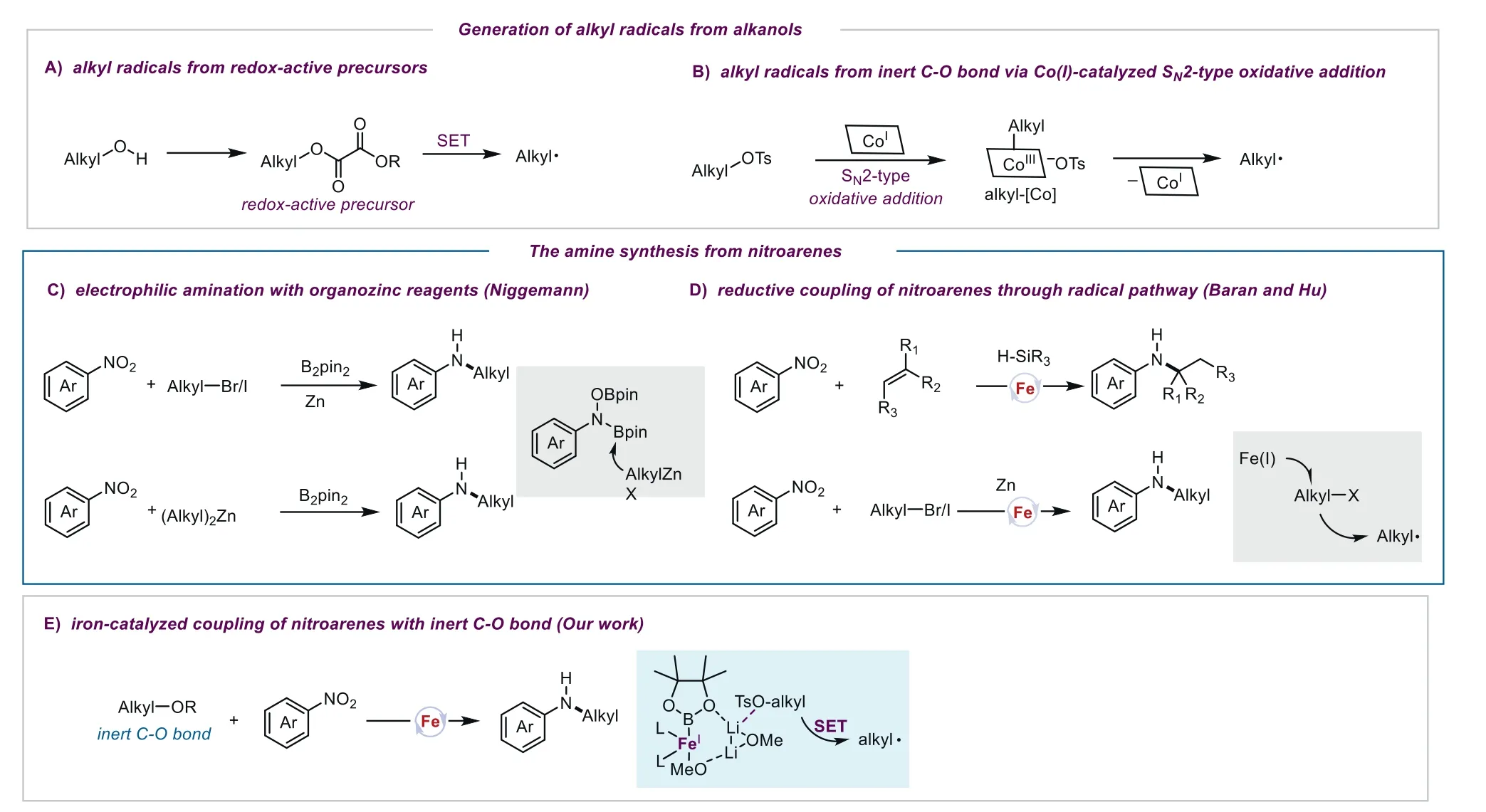
Fig.1. Methods for the generation of alkyl radicals and amines synthesis from nitroarenes.
Amines are the most important compounds in organic synthesis,widely existing in materials,pharmaceuticals and agrochemicals [22].The most widespread methods for amine synthesis are the Buchwald-Hartwig and Ullman-Ma C-N bond coupling reactions [23–25].It should be noted that these amination methods use anilines as the nitrogen sources,which are usually obtained by hydrogenation of nitroarenes.Consequently,the direct use of nitroarenes as starting materials for amine synthesis is more attractive due to its step economy and cost efficiency [26–41].In addition,in the classical C-N bond formation coupling reactions,the sensitive functional groups such as hydroxyl and thiol are not always compatible,which would readily react with electrophiles to afford alkylated or arylated products.In comparison,when nitroarenes are employed as coupling partners,such groups could be well tolerated.Recently,the Niggemann group developed a novel nucleophilic type of amination reaction of organozinc reagents with nitroarenes through a borane-promoted 1,2-migration (Fig.1C) [42,43].In 2015,the Baran group reported an elegant ironcatalyzed hydroamination protocol for amine synthesis from nitroarenes and alkenes,though this catalytic system is not suitable for primary alkylamines (Fig.1D) [44].Soon after,the Hu group employed zinc as a reductant to achieve the amination of alkyl bromides and alkyl iodides with nitroarenes [45],but the catalytic system was not suitable for inert substrates,such as alkyl chlorides and alkyl tosylates (Fig.1D).Therefore,it is necessary to develop more efficient catalytic systems for the inert C-O bonds cleavage to generate alkyl radicals.
Our recent studies demonstrated that theinsitugenerated ironboryl complex in the iron/borane reagent/alkoxide catalytic systems (such as Fe(OAc)2/B2pin2/MeOLi) have strong reducing properties,which could promote the transformation of the inert bonds[46,47].Inspired by these findings,we envisioned that,with such catalytic systems,inert alkyl tosylates may be reduced to generate alkyl radicalsviaa similar Fe/Li cation-assisted single electron transfer process [47],and nitroarenes may also be reduced to nitrosoarenes,and the resulting alkyl radicals could subsequently be trapped by nitrosobenzene to afford the amination products.Therefore,as a part of efforts in exploring the nature of iron catalysis [48–71],we investigated the iron-catalyzed amine synthesis using unreactive alcohol derivatives as substrates (Fig.1E).
Given these considerations,we set out to study this ironcatalyzed amination reaction by the treatment of 1-methyl-4-nitrobenzene1aand alkyl tosylate2bwith an iron catalyst in the presence oft-BuXphos.Reassuringly,the desired product 1 was obtained in 16% yield using MeONa as a base (Table 1,entry 1).After evaluation of various inorganic bases,MeOLi was found to promote this reaction smoothly with 25% yield (Table 1,entry 2;for details,see Supporting information).Various iron sources were further tested,and Fe(OTf)2exhibited good performance,providing1in 31% yield (Table 1,entries 3 and 4;for details,see Supporting information).Solvents proved to be important for this transformation,and CPME was a good choice,yielding the desired product in 38% yield (Table 1,entries 5 and 6;for details,see Support-ing information).Additionally,ligands were then checked,and the phosphine ligands facilitated this reaction well (Table 1,entries 7-9;for details,see Supporting information).Xantphos showed good reactivity,and 40% yield was obtained (Table 1,entry 8).To our delight,this reaction proceeded with good efficiency using bulky PAd3[72] as the ligand,providing1in 78% yield (Table 1,entry 10).It is reasoned that electron-rich PAd3ligand with bulky group may favor the generation of alkyl radical to facilitate the amination reaction.Switching CPME to MTBE and increasing the concentration afforded the best results,furnishing1in 86% isolated yield(Table 1,entries 11 and 12).Without the borane reagent,no desired product was observed (for details,see Supporting information).Other reductants such as Zn and Mn,were also examined,as anticipated,which could not promote this amination reaction (for details,see Supporting information).Furthermore,control experiments demonstrated the importance of iron catalyst and ligand,and only 12% yield was obtained in the absence of iron and ligand(for details,see Supporting information).The high-purity iron catalysts were examined as well,and comparable yields were provided(for details,see Supporting information).These results suggest that this transformation is promoted by the iron catalyst.
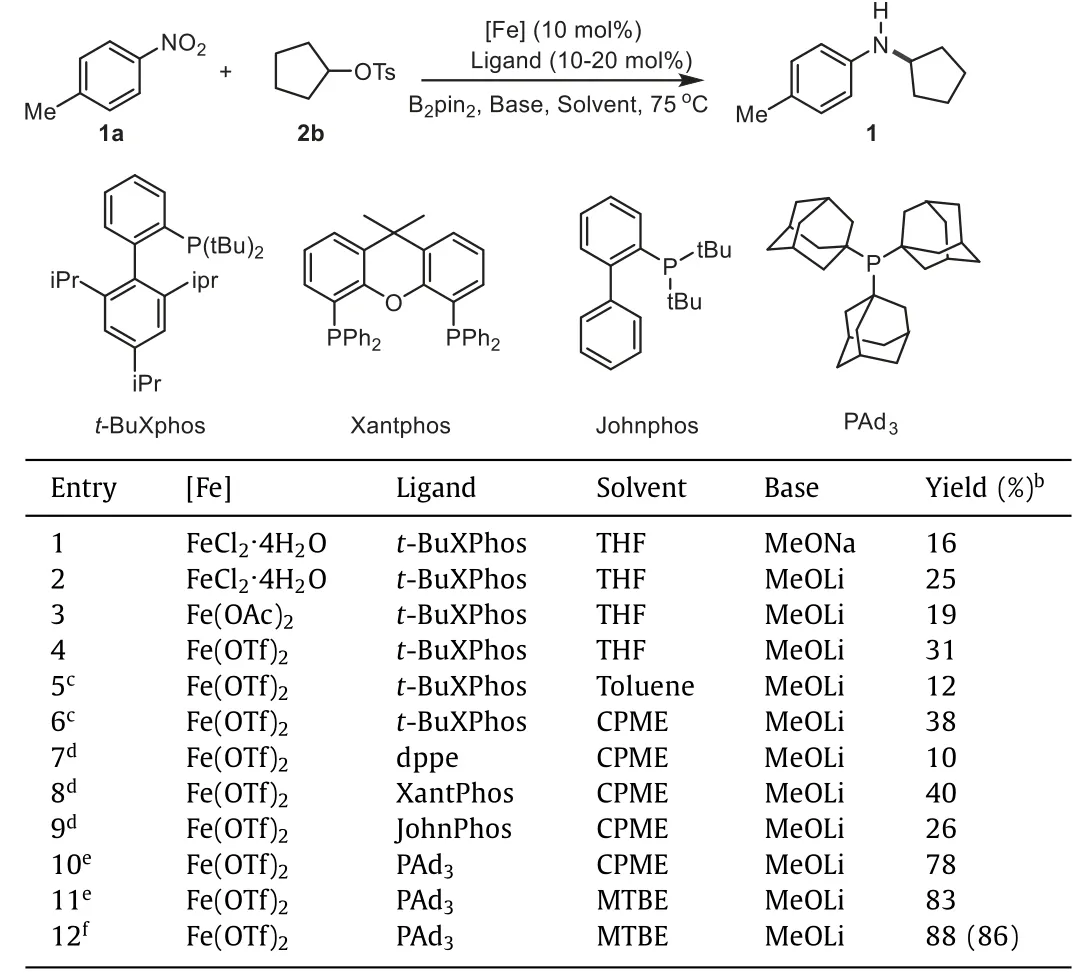
Table 1 Representative results for the optimization of iron-catalyzed reductive coupling of 1-methyl-4-nitrobenzene 1a with 2b.a
After the standard reaction condition was established,we examined the scope of this iron-catalyzed amination reaction.As shown in Scheme 1,this boron-promoted amination showed excellent functional group tolerance.Functional groups,such as,OMe,CN,OCF3,SMe,hydroxyl,F,Cl,Br,I,OTs,OTf,Bpin,carboxylate,morpholinyl,amide,CF3,alkenyl,alkynyl,and sulfone were all compatible with this catalytic system (4,6-19,31-34).Nitroarenes with electron-donating groups exhibited good reactivity,providing the corresponding products in moderate to good yields (3-5,75%-80%).The substrate with an electron-withdrawing group was also suitable for this transformation,giving rise to a moderate yield(10,76%).Importantly,the sensitive hydroxyl group usually readily reacts with alkyl halides under traditional C-N bond formation conditions,while it was well-tolerated in this protocol (9,71%).Substrates containing a halogen group or Bpin group proceeded this reaction smoothly (10-13,51%-76%;16,60%),providing a good chance for the downstream transformations.Nitroarenes bearing aπ-conjugated system performed this amination effi-ciently,furnishing the desired products in moderate yields (21-24,50%-70%).Moreover,the substrates bearing a heteroaromatic ring,such as pyridine,benzofuran,benzo[d]thiazole,benzo[d]oxazole,indole,and indazole were also demonstrated to be good substrates,delivering the corresponding products in reasonable yields (25-30,50%-71%).Subsequently,some alkyl tosylates were evaluated.Primary and secondary alkyl tosylates were good coupling partners,affording the amination products in moderate to good yields (31-53,45%-84%).Notably,alkyl tosylates with an alkenyl or alkynyl group could react well,providing the desired products in synthetically useful yields,while hydroboration of the unsaturated bonds did not occur (32,53%;33,54%).Alkyl tosylates bearing a functional group (such as sulfone and carboxylate) at the carbon chain underwent this amination smoothly,and moderate yields were obtained (34,58%;36,70%).

Scheme 1. Scope of the borane-promoted thiolation of (hetero)aryl sulfonyl chlorides.Reaction conditions: nitroarenes (0.2 mmol,1.0 equiv.),alkyl tosylates (0.4 mmol,2.0 equiv.),B2pin2 (0.55 mmol,2.75 equiv.),Fe(OTf)2 (0.02 mmol,0.1 equiv.),PAd3 (0.04 mmol,0.2 equiv.),MeOLi (1.6 mmol,8.0 equiv.),MTBE (0.8 mL),75 °C,15 h.a Alkyl tosylates (0.26 mmol,1.3 equiv.) were used.
To demonstrate the synthetic utilities of this reductive amination reaction,gram-scale syntheses were carried out under standard conditions.As shown in Scheme 2,when 1 g of1aor48bwas employed for this transformation,moderate yields of desired products were provided.In addition,some drugs and biomolecules were used as substrates to evaluate the applicability of this methodology.Nimesulide,a nonsteroidal anti-inflammatory drug,with a challenging sulfonamide group underwent this transformation smoothly,providing the corresponding product in synthetically valuable yields (54,30%).Nitrofen was used as an herbicide,and performed this amination well,affording the desired product55in 73% yield.Notably,the phenylalanine derivative with some sensitive groups such as ester and NHBoc,reacted well,and a good yield was obtained (56,73%).Substrates derived from cinchophen and gemfibrozil exhibited good performance,and good yields of the corresponding products were afforded (57,70%;61,70%).The telmisartan derivative carried out this amination effectively,delivering the coupling product58in 70% yield.Interestingly,cholesterol and stigmasterol derivatives showed excellent reactivity,furnishing the desired products in good yields (59,85%;60,84%);meanwhile,the alkenyl group on the alkyl tosylates could be reduced in this catalytic system.
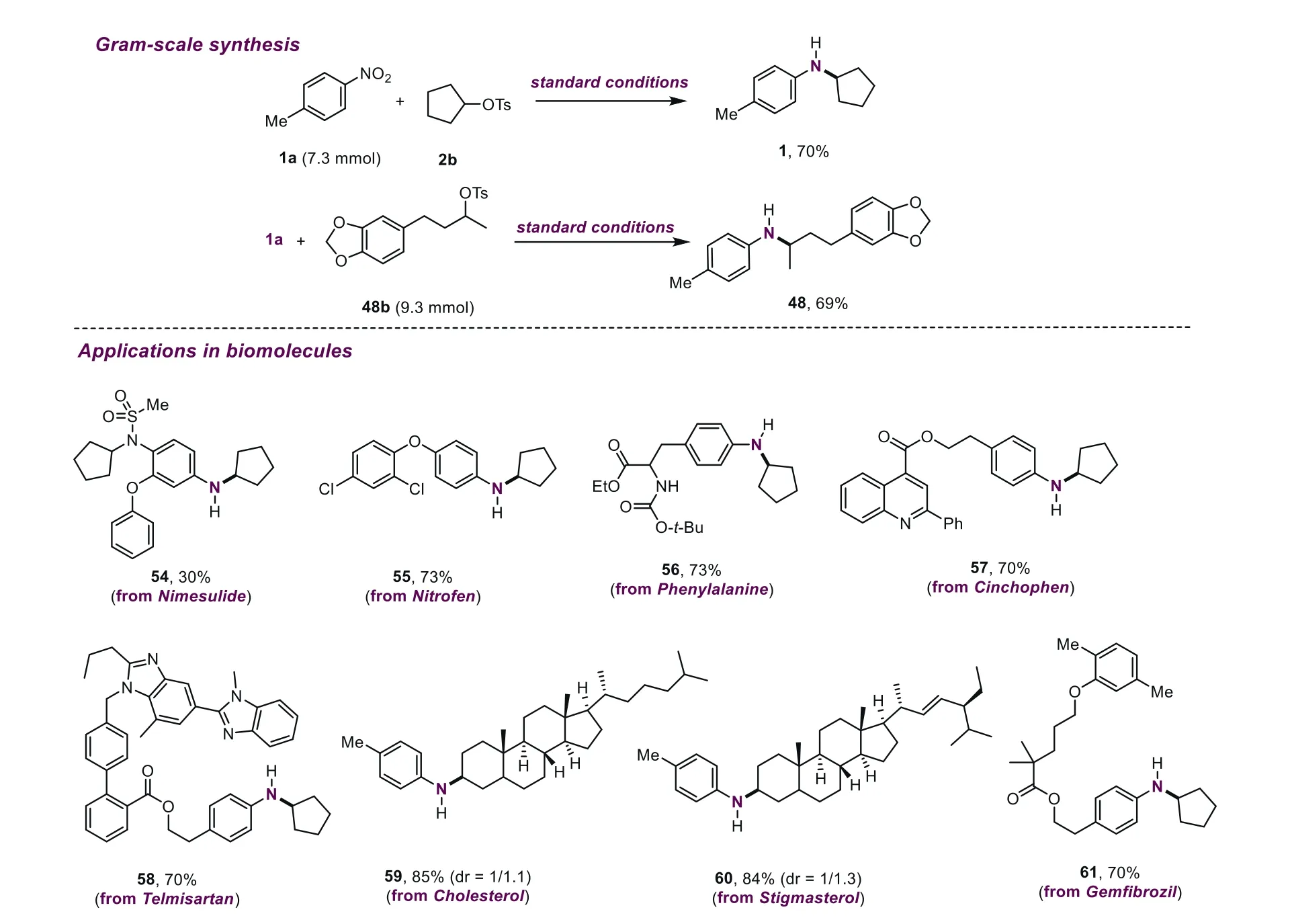
Scheme 2. Gram-scale synthesis and late-stage functionalization of biomolecules.
Subsequently,some experiments were performed to shed light on the mechanism of this iron-catalyzed amination reaction.First,this transformation could be completely depressed,when a radical scavenger TEMPO (200 mol%) was added under the standard reaction conditions (for details,see Supporting information).Moreover,a radical clock experiment was performed,and the major ring-opening product63was obtained (Scheme 3A).These results suggest that the radical pathway is dominant in this amination reaction,and the SN2 pathway is also involved.Subsequently,we attempted to investigate the possible intermediates in this transformation.In our catalytic system,nitrosoarene64a-1could be isolated,and according to the literature [27,29,30],some potential intermediates derived from nitrobenzene64awere evaluated.As shown in Table S16 (Supporting information),nitrosoarene64a-1andN-phenyl hydroxylamine64a-2could provide moderate yields,while other compounds performed this transformation with lower efficiency (for details,see Supporting information),suggesting that nitrosoarene andN-aryl hydroxylamines were the plausible intermediates in the iron-catalyzed reductive amination.In our recent work [46,47],it was found that the iron/B2pin2/MeOLi catalytic systems could allow the high-valent iron species to be reduced to deliver the low-valent iron(I)-boryl species.Therefore,a dppeligated iron(I)-Cl complex [73] were synthesized,which could indeed promote this amination,affording the desired product in moderate yields (Scheme 3B).These outcomes suggest that iron(I)species might be participated in the catalytic cycle.
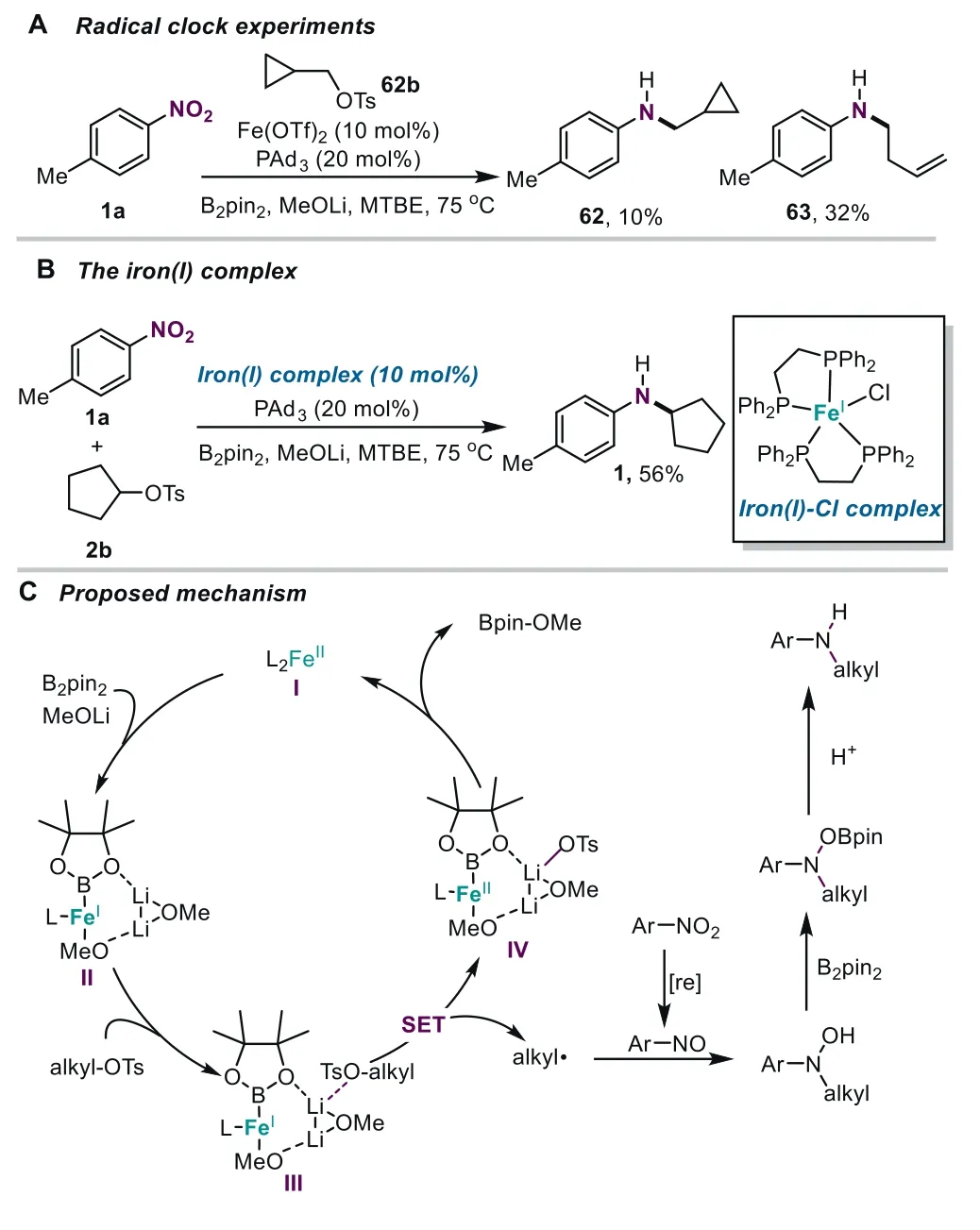
Scheme 3. Mechanistic studies.
On the basis of these mechanistic studies,a plausible mechanism is proposed (Scheme 3C).First,Fe(II) species was reduced by B2pin2with the assistance of MeOLi to form a low-valent iron(I)-boryl complex [47].The resulting intermediateIIcould coordinate with alkyl-OTs to produce alkyl radicalsviaa Li cation-assisted single electron transfer pathway.Additionally,nitroarene could be reduced to form nitrosoarene in the iron/borane reagent/alkoxide catalytic systems [45].Subsequently,theinsitugenerated alkyl radical was trapped by nitrosoarene to afford theN-aryl hydroxylamine.Finally,the resultingN-aryl hydroxylamine reacted with B2pin2and underwent protonation to afford the coupling amination product [44].
In summary,we have developed an efficient method for the activation of inert alkyl C-O bonds to generate alkyl radicals through an iron/borane reagent/alkoxide catalytic system.This transformation exhibits excellent functional group compatibility and latestage functionalization of bioactive molecules,thus offering good opportunities for applications in drug discovery and development.Mechanistic studies suggest that the iron/B2pin2catalytic system can induce the cleavage of inert alkyl C-O bonds to generate alkyl radicals,and the amination reaction proceeds through the alkyl radical addition pathway.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank for the financial support from National Natural Science Foundation of China (Nos.22271031,22201026),Natural Science Foundation of Chongqing (No.CSTB2022NSCQ-MSX1065),Chongqing Postdoctoral Science Foundation (No.cstc2020jcyjbshX0052),Medical Imaging Key Laboratory of Sichuan Province(Nos.MIKL202201 and MIKL202202),Affiliated Hospital of North Sichuan Medical College (No.2022JB001),and Youth Project of Science and Technology Research Program of Chongqing Education Commission of China (No.KJQN201900112).We also thank Analytical and Testing Center of Chongqing University for assistance with NMR spectrum analysis.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108303.
 Chinese Chemical Letters2023年12期
Chinese Chemical Letters2023年12期
- Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers