
Al 原子的替位掺杂与表面吸附对BiVO4 (010)晶面光电催化分解水析氧性能的影响*
2023-02-18 06:38李秋红马小雪潘靖
物理学报 2023年2期
李秋红 马小雪 潘靖
(扬州大学物理科学与技术学院,扬州 225002)
太阳能光电催化分解水制氢气和氧气是获得可再生能源的可行方案之一,利用密度泛函理论计算,对比了替位掺杂和表面吸附过渡金属Al 原子对BiVO4 (010)晶面析氧(OER)性能的影响.结果表明,两种方式都能有效调控BiVO4 的电子结构进而调节其OER 性能,而表面吸附因能改善BiVO4 的导电性和光吸收,降低电子-空穴复合,增强OER 过程中活性位点与含氧中间体之间的相互作用,降低决速步的过电势,被认为是提高(010)晶面析氧性能的有效手段.本工作为设计高效的二维半导体析氧反应的光催化剂提供了重要参考.
1 引言
太阳能光电催化(PEC)分解水制取氢气和氧气作为一项新型技术,是实现清洁可再生能源的有效手段[1−3].寻找和设计高效稳定的光电催化剂是实现光催化分解水工业应用的关键,理想的光电催化剂须满足: 具有匹配的带边位置即价带顶(VBM)和导带底(CBM)跨过水的氧化还原势,理想的禁带宽度同时能很好地吸收可见光,良好的载流子迁移效率,较低的电子空穴复合率,高稳定性等等[4−8].其中,n 型半导体材料单斜钒酸铋(ms–BiVO4)禁带宽度为2.4 eV,光吸收范围300—550 nm,能有效吸收可见光;同时具有较好的抗氧化性,在自然界中普遍存在.更值得关注的是BiVO4的导带边跨过了析氧反应(OER)的过电势,是最具潜力的析氧反应催化剂候选材料之一[9−11].众所周知,BiVO4具有多个暴露晶面,其中(010)是公认的析氧反应较好的晶面[12,13].然而,单斜钒酸铋仍然面临着电子空穴复合率较高,载流子迁移率较差的问题;同时,在可见光区域的光吸收仍有待进一步提高.为了改善ms–BiVO4析氧性能,研究者进行了很多尝试,其中,单斜钒酸铋中掺杂Se,W,S,F,Mo,Ir 等金属或非金属是可行方案之一[14−17],比如Ullah 等[17]在单斜钒酸铋中掺入非金属Se 原子,能有效提高载流子迁移率;Maheskumar 等[18]利用S 掺杂从结构、光学、电子方面提高钒酸铋的光催化性能;Zhao 等[19]引入金属W 掺杂,在原活性位点Bi 的基础上增加了新的活性位点V,并使W 成为新的水吸附位点,增加了活性位点数量.值得注意的是过渡金属铝具有良好的导电性,Ma 等[20]发现Al 掺杂能有效改变NiMoO4的电子结构,进而改善其析氢析氧性能.本工作分别采用Al 原子掺杂和表面Al 原子吸附两种方式来调节ms–BiVO4(010)面的结构,并从电子结构、导电性、光吸收、析氧过程中含氧中间体的吸附能以及决速步过电势出发,对比两种方式对钒酸铋析氧性能的影响,计算结果表明: Al 原子的替位掺杂和表面吸附均能改变BiVO4的电子结构,降低禁带宽度,增强对可见光的光吸收,但替位掺杂未能有效改善(010)面析氧性能.而Al 原子的表面吸附,能有效降低光生空穴的有效质量,促进了光生空穴迁移;电子和空穴有效质量比的计算发现表面吸附降低了电子-空穴复合率;吸附能的计算发现Al 的介入增强了析氧过程中含氧中间体与表面的相互作用,有效降低决速步的过电势,提升了表面的析氧活性.
2 理论和计算方法
本次工作中所有的计算都是在密度泛函理论(DFT)框架下[21,22],应用计算软件VASP 进行的.以平面投影缀加波 (PAW)赝势描述离子实和价电子之间的相互作用[23,24].分别采用广义梯度近似(GGA)的PBE 方案处理电子间相互作用的交换关联能.其中,平面波截断能为400 eV.几何优化和态密度在对布里渊区的积分计算采用K点网格分别为7×7×1 和11×11×1 的 Monkorst-Park方案.结构优化中,原子位置、晶格常数都进行了优化,能量收敛标准为1.0 × 10–5eV,原子力收敛标准为0.01 eV/Å.
如图1(a)所示,单斜BiVO4采用空间群I2/a体系,包括V,Bi,O 三种原子,由VO4四面体和BiO8十二面体共同构成.这里,以单斜钒酸铋(010)晶面为研究对象,为避免周期性作用,在(010)表面增加20 Å的真空层.如图1(c)所示,选取两层1×1 的超胞,共48 个原子,包含8 个Bi 原子,8 个V 原子,32 个O 原子.其中表面Bi 是析氧反应的活性位点[25],为了更好地考察Al 掺杂和吸附对活性位点的影响,如图1(d)所示给出了Al 原子替位掺杂V 的情况,活性位点周围有3 个V 位点与其邻近,这里将一个Al 原子分别替换活性位点附近本征的V 原子;图1(e)给出了Al 原子表面吸附的情况,Al 吸附在表面V 的顶位,并位于两个O 原子之间.
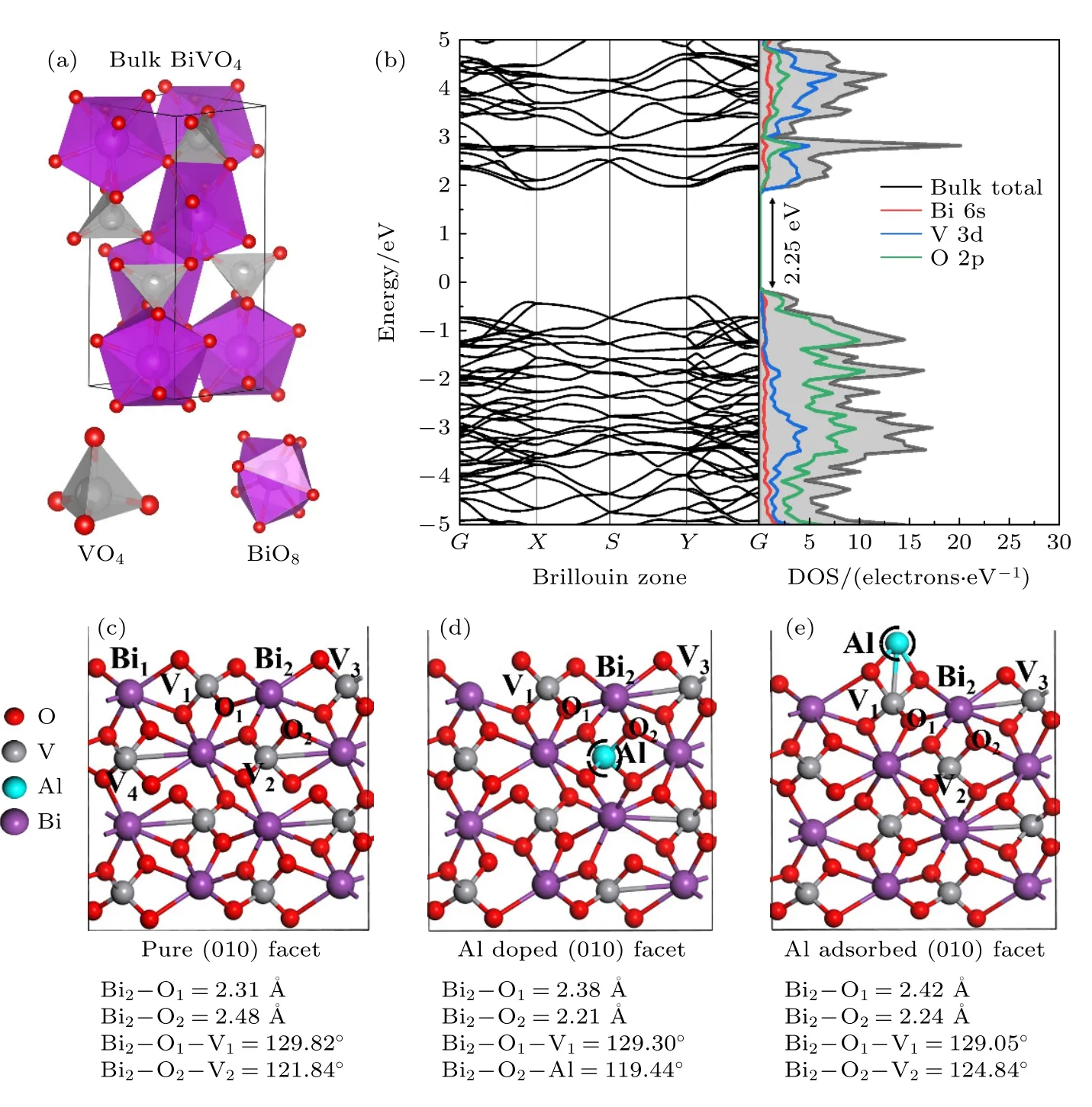
图1 (a) ms-BiVO4 块体结构侧视图;(b) 块体钒酸铋的能带结构与态密度;(c) BiVO4 (010)面结构的侧视图;(d) Al 替代V 位点的侧视图;(e) Al 原子吸附在BiVO4 (010)晶面的侧视图;BiVO4 (010)晶面共48 个原子,包括8 个Bi (紫色)、8 个V (灰色)和32 个O (红色)原子Fig.1.(a) The side view of bulk ms-BiVO4; (b) band structure and PDOS of bulk BiVO4; the side views of (c) pristine,(d) Al doped and (e) Al adsorbed BiVO4 (010) facets.There are 48 atoms in BiVO4 (010) facet including 8 Bi (purple),8 V (gray),and 32 O (red) atoms.
3 计算结果与讨论
3.1 Al 原子替位掺杂和表面吸附对BiVO4(010)晶面结构的影响
为进一步提高BiVO4(010)面析氧性能,采取替位掺杂和表面吸附过渡金属Al 原子这两种方式.首先,对单斜钒酸铋块体模型进行了结构优化,GGA–PBE 计算得到的晶格常数分别为a=5.04 Å,b=5.27 Å,c=11.89 Å,与实验值a=5.10 Å,b=5.17 Å,c=11.70 Å基本一致[26,27],其能带结构和原子的分波态密度如图1(b)所示,其中,价带顶主要由O 2p 轨道组成,导带底主要由O 2p 和V 3d 轨道组成,表明电子跃迁途径主要是从价带顶的O 2p 轨道跃迁到导带底V 3d 轨道,这与文献[28]报道结果基本一致,计算得到其禁带宽度为2.25 eV,与GGA+U,HSE06 方法所得值2.147,2.8 eV 相比,更接近于实验值2.45 eV[13,25,29],证明GGA-PBE 方法对于计算钒酸铋结构具有较好的可靠性.
BiVO4(010)表面Bi 是析氧反应活性位点[25],如图1(c)所示,表面包括Bi1和Bi2两个潜在的水吸附位点,这里选取体系中心的Bi2作为活性位点,因为有相对更多的V 位点与其相邻,可作为掺杂位点被Al 替代.从结构分析,发现Bi2与邻近的O 的键长以及与周围原子形成的键角各不相同,其中Bi2—O1键长2.31 Å,Bi2—O2键长2.48 Å,Bi2—O1—V1键角129.82°,Bi2—O2—V2键角121.84°,说明其结构具有不对称性.而掺杂和表面修饰将进一步破环其对称性,使结构发生变化,进而改变其电子结构,对体系的催化活性产生影响.为了更好考察替位掺杂与表面吸附对BiVO4(010)晶面OER 性能的直接影响,本文给出了Al 原子近邻掺杂和近邻吸附的情况.如图1(c)所示,分别考察V1,V2,V3,V4作为掺杂位点的情况,结果发现当V1,V3被Al 原子替位掺杂时,优化过程中表面层发生断裂,结构难以保持稳定.分析认为: 根据鲍林标度,V 的电负性为1.6,Al 的电负性为1.5,由于电负性的变化使得Al 替代V1后对周围原子的吸引能力变弱,同时V1原子半径为1.92 Å,Al 的原子半径为1.82 Å,在优化过程中掺杂元素Al 与周围原子重新成键,键长、键角发生改变,体系趋于能量最低状态时构型发生改变,难以保持稳定,因此这两种掺杂被自然淘汰.图1(d)展示了V2原子作为替代位点被Al 原子掺杂的结构优化模型,其 中,Bi2—O1键长变为2.38 Å,Bi2—O2键 长2.21 Å,Bi2—O1—V1键角129.30°,Bi2—O2—Al键角119.44°.对于表面吸附而言,在Bi2近邻处引入了Al 原子吸附,由于表面V 和O 的共同作用,Al 被吸附到了V 顶位,形成了Al—O 离子键,键长1.90 Å,Al 与V 之间形成了金属键,键长2.67 Å.而Bi2与周围的O 原子的键长以及键角进一步变化,Bi2—O1为2.42 Å,Bi2—O2为2.24 Å,Bi2—O1—V1为129.05°,Bi2—O2—V2为124.84°.为了考察掺杂和表面修饰难易程度以及体系的稳定性,计算了体系的形成能[30]:
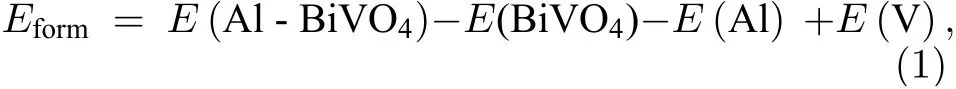
其中,E(Al-BiVO4)为铝修饰后体系的总能量、E(BiVO4)为钒酸铋(010)面的总能量、E(Al)为铝的单质块体能量、E(V)为钒单质块体的能量,计算得到Al 掺杂V2位点体系的形成能Eform为–6.32 eV,Al 替换V4后体系形成能为–6.3 eV,但由于V4位置的掺杂离活性位点较远,对活性位点的催化活性影响较小,因此在后面的讨论中,被省略.Al 原子表面吸附体系形成能Eform为–2.92 eV,形成能为负值,表明无论是替位掺杂还是表面吸附在(010)晶面都容易实现,并且体系保持稳定.
3.2 Al 原子替位掺杂和表面吸附对BiVO4(010)晶面电子结构和导电性的影响
结构的变化必然引起BiVO4电子结构的变化,图2(a)给出了BiVO4(010)晶面的总态密度与分波态密度,其禁带宽度Eg为2.22 eV,导带底由V 3d,O 2p 以及Bi 6p 轨道组成,价带顶主要由O 2p 轨道组成,与插图中给出的电荷密度一致,导带底电荷集中在V 原子周围,价带顶电荷集中在O 原子周围.Al 的替位掺杂使BiVO4电子结构发生了改变,如图2(b)所示,价带处O 的2p 轨道向费米能级移动,减小了BiVO4的禁带宽度,Eg降低至2.03 eV,禁带宽度的降低表明电子跃迁所需要的最小能量下降,更有利于电子从O 2p 轨道向V 3d 轨道跃迁,有利于光吸收的增强;同时电荷也发生了转移,电荷密度图表明Al 掺杂减少了表面的电荷分布.图2(c)发现Al 原子吸附降低了BiVO4禁带宽度,Eg降为2.02 eV,同时,在导带与价带之间的0.71—0.87 eV 处出现了局域的电子态,这主要来自于Al 3 s,V 3d 和少数O 2p 之间的杂化;禁带宽度的降低有利于热激发,杂质能级的出现有利于电导率的增强,同时会成为电子-空穴的复合中心,但对于浅能级而言,更易于载流子的产生.对光催化而言,宽禁带(带隙为2.02 eV)中的杂质能级(其价带顶到杂质能级的间隙为0.87 eV,杂质能级到导带底的间隙为0.71 eV)能扩展光的吸收范围,以增加光生载流子,进而增大光的利用效率.因此,虽然其可能成为复合中心,但总体上还是增大光催化效能.其对于电荷的跃迁起到了很好的辅助作用: 光生电荷从价带顶跃迁到杂质能级,再从杂质能级跃迁到导带底[31],与图2(a),(b)中未修饰以及Al 替位掺杂的情况相比,电荷跃迁变得更加容易,这在一定程度上更有利于光子的吸收,加速光生电荷的跃迁.此外,从电荷密度图可以清楚地看到杂质能级处,电荷集中在表面的Al 以及近邻的O,V 原子周围,表面电荷逐渐增强.
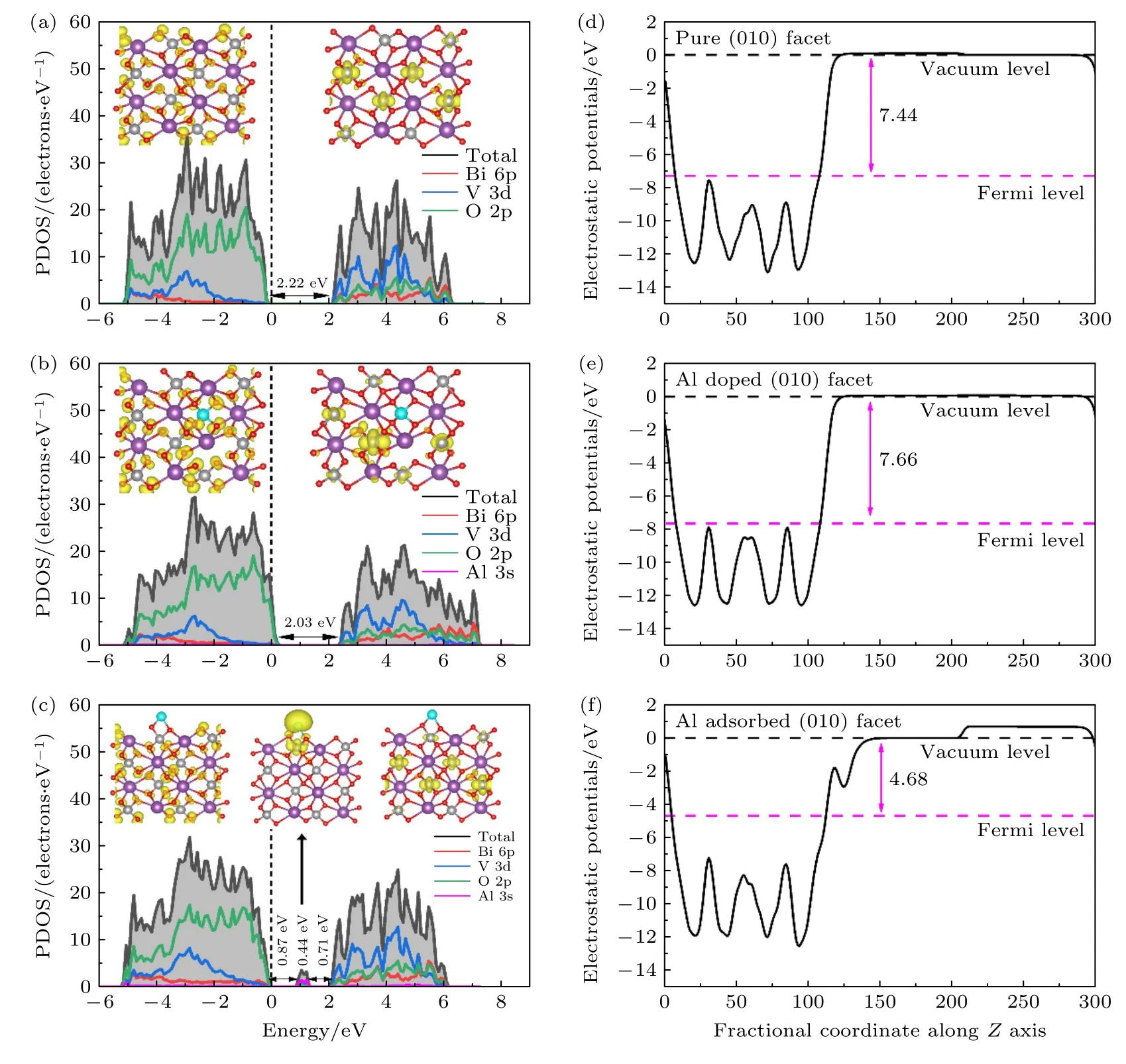
图2 (a) 原始的、(b) Al 替位掺杂和(c) 表面吸附的BiVO4 (010)晶面总态密度和分波态密度;插图是导带底和价带顶处电荷密度图,费米能级设置为零;(d) 原始的、(e) Al 替位掺杂和(f) 表面吸附BiVO4 (010)晶表面沿 z 轴的平均静电势Fig.2.The total and partial density of states of (a) pure,(b) Al substitutional doped and (c) surface adsorbed BiVO4 (010) surfaces;the inset is the charge density of VBM and CBM,the Fermi level is set to zero.Average electrostatic potentials along the z axis of (d) pure,(e) Al substitutional doped and (f) surface adsorbed BiVO4 (010) surfaces.
图2(d)—(f)给出了体系的功函数,所谓功函数是指载流子从固体内部逸出到真空层所需要的最小能量,具体表现为真空能级与费米能级之差[32],计算得到 BiVO4(010)面的功函数W⊥为7.44 eV,Al 替位掺杂增大了功函数,变为7.66 eV,而Al 的表面吸附降低功函数为4.68 eV.这与电荷密度分布情况一致: 从图2(c)可以看出,杂质能级处的电荷集中在表面Al 以及周围原子之上,表面电荷增多,是功函数降低的原因;对于原始的BiVO4(010)面而言,导带底处,电荷集中在V 原子周围(参见图2(a)中的插图),Al 替代内部的V2原子后,电荷重新分布,但新引入的Al 原子并未带来新的电荷分布,而对电子跃迁提供主要贡献的V 原子浓度降低(参见图2(b)中的插图),这在一定程度上降低了导带处电荷贡献,是导致功函数升高的原因.
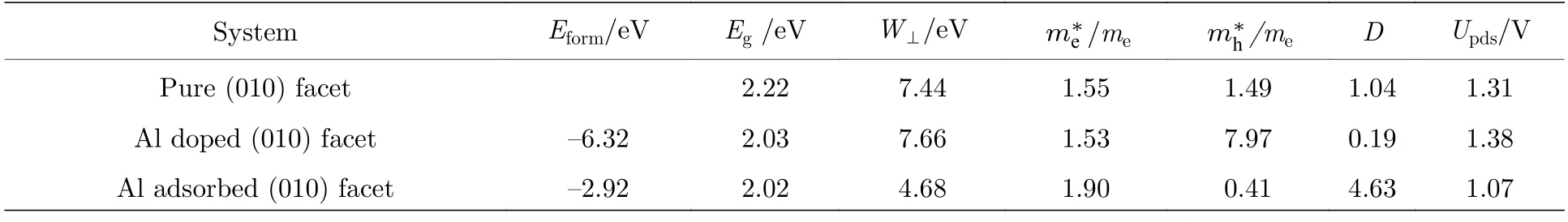
表1 原始的及Al 原子替位掺杂和表面吸附的钒酸铋(010)晶面的形成能、禁带宽度、功函数、电子有效质量、空穴有效质量、电子-空穴有效质量比、决速步过电势Table 1.The formation energy,band gap,the work function,effective mass of electron,effective mass of hole and relative ratio of the effective masses,the potential of the rate determining step of pure,Al substitutional doped and surface adsorbed BiVO4 (010) surfaces.
在半导体光电催化材料中,电子和空穴有效质量比决定了光生载流子的分离速率,有效质量比由以下公式定义:

1 和D值之间的差值越大,光生电子与空穴的扩散速率差异越大,这意味着光催化剂的电子-空穴的复合速率越低[16],原始的(010)晶面的有效质量比为1.04,表明光生电子和空穴容易复合,Al 替位掺杂降低了(010)面的有效质量比,为0.19,而Al 原子表面吸附极大的提高了有效质量比,为4.63,这表明Al 原子的表面吸附很大程度上降低了光生电子-空穴的复合速率.
3.3 Al 原子替位掺杂和表面吸附对BiVO4(010)晶面光吸收的影响
图3(a)给出了光吸收谱图,原始的BiVO4(010)面的吸收峰位于207.7 nm 处,Al 替位掺杂和表面吸附使BiVO4吸收峰红移,分别位于208.5 nm和210.5 nm 处,在300—550 nm 处,Al 替位掺杂和表面吸附的BiVO4光吸收明显增加;同时发现,Al 原子表面吸附体系光吸收曲线在362 nm 处出现一个小的吸收峰,进一步增强了可见光区域的光吸收.为了清晰地表征禁带宽度的变化,可以利用改进的Tauc 方程进行描述[29,34],其中F(R)表示Kubelka–Munk 函数,hν表示光子能量,Eg为禁带宽度,A表示比例常数,指数n表示跃迁的性质(n=2 为直接半导体,n=1/2 为间接半导体).图3(b)中,切线截距即为钒酸铋禁带宽度,原始的BiVO4(010)面和Al 原子替位掺杂的禁带宽度分别为2.22 eV 和2.03 eV,与之相比,Al 原子表面吸附使(010)面具有两个小吸收峰,切线与横轴相交获得两个截距能量,第1 个峰的截距能量为2.02 eV 与禁带宽度有关,第2 个峰的截距能量为2.46 eV,二者之间的差值为0.44 eV,与局域态的能量范围有关(参见图2(c)).实际上,光吸收谱与导带和价带之间的电子跃迁密切相关,对于原始的(010)面和Al 替位掺杂的(010)面而言,光生电子从价带顶O 的2p 轨道跃迁到导带底V 3d,Bi 6p 和O 2p 轨道(见图2(a)和(b));而对Al 吸附的(010)面,增加了中间态Al 的3 s 轨道,由于局域电子态的存在,电子跃迁不仅包括价带顶到导带底的跃迁,还包括从局域态即Al 3s,V 3d,O 2p 轨道到导带底的跃迁(见图2(c))[31].这表明局域电子态不仅有利于光子的吸收,而且提高了光生电子的跃迁效率.
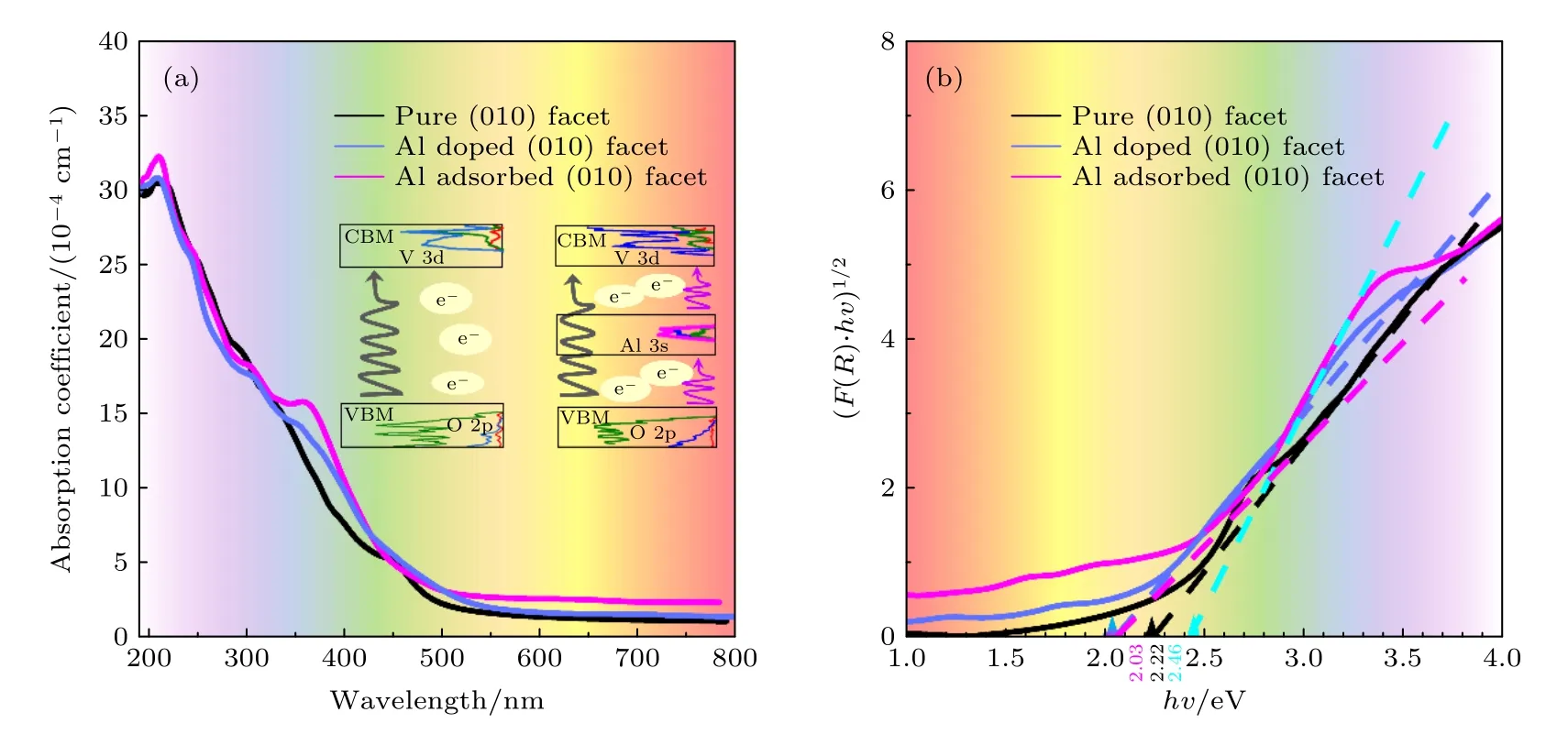
图3 原始的、Al 替位掺杂和表面吸附BiVO4 的(010)晶面的(a)光吸收谱图;(b) (F(R)·hν)1/2 与光子能量关系图像Fig.3.(a) The calculated absorption coefficient and (b) the (F(R)·hν)1/2 with the change of photon energy in pure,Al substitutional doped and surface adsorbed BiVO4 (010) surfaces.
3.4 Al 原子替位掺杂和表面吸附对BiVO4(010)晶面OER 性能的影响
析氧反应(OER)包含了4 个质子耦合电子转移步,相较于析氢反应(HER)两步电子的转移,其动力学过程更为缓慢.对于原始的钒酸铋(010)表面OER 反应而言,第一步,水吸附在表面层,在光生空穴的作用下,所吸附的H2Oads失去一个H+,形成HOads,吸附在(010)表面;第二步,HOads失去一个H+形成Oads,吸附在表面;第三步,另一个H2O 吸附于表面,在光生空穴的作用失去一个H+,形成HOOads吸附;最终,在偏电压的作用,两个具有亲电性的氧原子成键,从表面脱离,形成O2,整个析氧反应过程如(3)—(6)式:

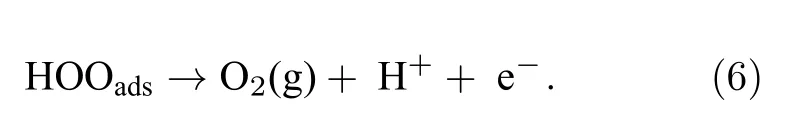
过程中,通过析氧反应中间体H2Oads,HOads,Oads和HOOads吸附能来考察动力学过程的难易程度,根据公式Eads=Emolecule+surface–Emolecule–Esurface(其中Emolecule+surface,Emolecule,Esurface分别表示吸附后体系的总能量,吸附物的能量,初始的(010)面的能量)计算各中间态的吸附能,如图4所示给出了OER 的吸附过程,对于Al 替位掺杂而言,水吸附于活性位点Bi2的顶位,吸附能相较于原始(010)面的–0.64 eV 变为–0.70 eV,体系结构更加稳定,吸附能负值越负说明水越容易吸附;对于Al 表面吸附的(010)表面而言,水分子更加倾向于吸附在表面的Al 与Bi 的桥位上,其中,Bi—O 键长为3.12 Å,大于Al—O 键长(2.03 Å),由于Al 原子吸附使BiVO4电荷重排,更多的电荷集中到了表面的Al 原子周围,水吸附的吸附能为–0.75 eV;相较于替位掺杂,表面吸附更容易进行水吸附.据此可推测,Al 原子本身是否具有吸附含氧中间体的能力.进一步以Al 作为活性位点,进行了水吸附,结果发现,水吸附在Al 的顶位,其中Al—O 键长为2.01 Å,水的吸附能进一步下降为–0.78 eV.
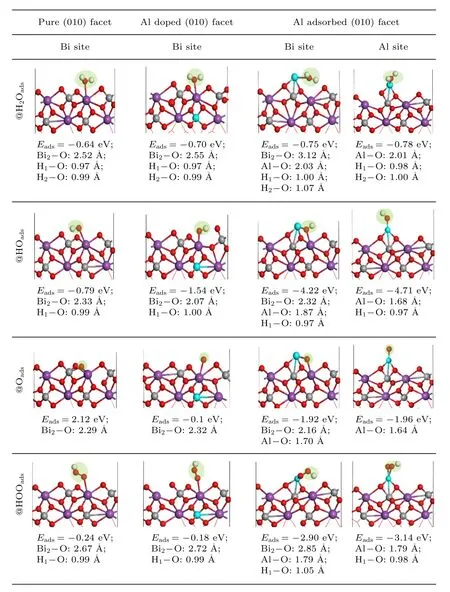
图4 OER 四电子步过程中含氧中间体H2Oads,HOads,Oads 和HOOads 吸附在原始的、Al 替位掺杂和表面吸附的BiVO4 (010)表面以Bi 或Al 为活性位点的吸附结构和吸附能.“–”和“@”符号分别表示(010)面上的键和吸附状态、吸附能与键长的统一单位为eV 和ÅFig.4.The adsorbed structure and adsorbed energies of the oxygenated intermediates of H2Oads,HOads,Oads and HOOads adsorbed on pure,Al substitutional doped and surface adsorbed BiVO4 (010) surfaces during the four steps of OER,where Bi and Al respectively act as active site.The “–”,and “@” signs stand for bond,and adsorption state on the surface,the unity units of adsorption energy and bond length are eV and Å,respectively.
同样的变化也发生在含氧中间体的其他吸附过程 中,在原始的BiVO4体 系中 HOads,Oads和HOOads吸附能分别为–0.79,2.12 以及–0.24 eV,Al 的替位掺杂使其降为–1.54,–0.1 和–0.18 eV.与其相比,Al 表面吸附更有利于中间过程的进行,而相对于以Bi 为活性位点而言,以Al 原子作为活性位点进行HOads,Oads和 HOOads吸附时,吸附物中的Al—O 键长大大降低,由Bi 为活性位点时的1.87,1.70,1.79Å 变为1.68,1.64 和 1.79 Å,键长变短意味着电荷从吸附位点向各吸附态转移变得更容易,活性位点与O 之间的相互作用增强,吸附能也由–4.22,–1.92,-2.90 eV 降为–4.71,–1.96 和–3.14 eV.Al 的表面修饰使BiVO4展现出了更好的OER 性能,不仅体现在与中间含氧体的相互作用,同时可发现,过程中由于表面吸附原子Al 的引入,使Bi 位点的活性得到极大提升.
图5 所示为计算的BiVO4(010)表面OER 过程自由能.自由能被定义为: ΔG=ΔE+ΔZPE–TΔS,其中ΔE指每个电子转移步的反应能,ΔZPE 为零点能量的变化量,ΔS为熵贡献的变化量.一个水分解反应H2O→1/2O2+H2的进行需要2.46 V 的电压,最小自由能应使两个水分子在4.92 V 的电压下分解,将最高自由能变化量ΔGmax对应的反应步骤定义为决速步(the rate determining step),过电势Upds为(ΔGmax–1.23) V.对于原始(010)面而言,决速步为第一步,即吸附H2O形成HOads,吸附位点为Bi,过电势Upds=1.31 V;引入Al 替位掺杂和表面吸附后,Oads和HOOads吸附变得更加容易,具有了更负的吸附能(参见图4),但这在一定程度上可能不利于OER 中间体的转化,使决速步从第一步变为第三步,即HOOads形成过程,其中Al 的替位掺杂,过电势为1.38 V;而在Al 表面吸附的BiVO4中,由于表面Al 原子在OER 过程中对活性位点Bi 的正向促进(具体参见图4),使Bi 位点活性得到提升,在双位点的共同作用下,过电势降低为1.07 V,有利于OER 反应的发生.

图5 OER 四个电子步在U=0,pH=0,T=298 K 下自由能台阶图 (a)原始BiVO4 (010)晶面;(b) Al 替位掺杂(010)晶面;(c) 表面吸附(010)晶面,Upds 表示决速步的过电势Fig.5.Free energy profiles of OER on (a) pure BiVO4 (010) facet;(b) Al doped (010) facet and (c) Al adsorbed (010) facet at U=0,pH=0,T=298 K.Upds represents the potential of the rate determining step.
4 结论
利用密度泛函理论(DFT)深入研究Al 原子替位掺杂与表面吸附对BiVO4(010)晶面光电催化分解水析氧性能的影响.结果表明,Al 原子替位掺杂和表面吸附能有效调控BiVO4的电子结构,从而调节其表面的催化活性,而Al 原子表面吸附展现出更优的OER 催化性能,是提高(010)晶面析氧反应的有效手段,主要表现在: 1)诱导的局域电子态以及禁带宽度的降低有利于电子跃迁,提高了可见光区域的光吸收和导电性;2)具有更低的空穴有效质量,能有效增强从阳极表面到电解质表面的转移能力,其中电子和空穴的有效质量比与1 差值增大,有效降低了电子-空穴的复合率;3)增强了活性位点与含氧中间体之间的相互作用,有效降低决速步的过电势.引入表面吸附过渡金属Al 是提高BiVO4的光电催化分解水析氧性能的有效手段,并且对于设计高效的二维半导体光催化剂提供了重要的参考.
猜你喜欢
火炸药学报(2022年5期)2022-11-04
装备维修技术(2021年36期)2021-10-25
弹箭与制导学报(2021年3期)2021-07-30
陕西科技大学学报(2020年6期)2020-11-25
无线互联科技(2019年15期)2019-11-07
物理实验(2019年7期)2019-08-06
网印工业(2019年4期)2019-05-21
重型机械(2019年2期)2019-04-28
航空材料学报(2019年2期)2019-04-15
物理学报(2018年22期)2018-12-18
