
Apolipoprotein A1 suppresses the hypoxia-induced angiogenesis of human retinal endothelial cells by targeting PlGF
2023-02-11 08:59:04JieHuZhuTingChenKunYiSuYuLianLinLuAnDiNaHu
Jie Hu, Zhu-Ting Chen, Kun-Yi Su, Yu Lian, Lin Lu, An-Di-Na Hu
State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat‐sen University, Guangdong Provincial Key Laboratory of Ophthalmology and Visual Science, Guangzhou 510060, Guangdong Province, China
Abstract
● KEYWORDS: apolipoprotein A1; retinal neovascularization;placental growth factor; MEK/ERK signaling pathway
INTRODUCTION
Retinal neovascularization is an essential pathological process involved in various retinal diseases, including proliferative diabetic retinopathy (PDR), retinal vein occlusion(RVO), and retinopathy of prematurity (ROP)[1]. Retinal neovascularization, the essential pathological process involved in PDR, causes bleeding, leakage, and even scar formation, leading to severe visual impairment[2‐3]. Therefore,it is necessary to investigate effective treatments for retinal neovascularization.
In the clinic, we observed that a distinct cohort of diabetic individuals were somehow “immune” to the onset of PDR,the retinal neovascularization stage. Then, we found that there was a significant association between decreased serum apolipoprotein A1 (apoA1) levels and PDR[4]. ApoA1 is the major apoA protein and accounts for approximately 70% of the total protein mass of high‐density lipoproteins[5]. ApoA1 has anti‐inflammatory, antioxidant and intraretinal reverse transport effects on lipids[6‐8]. The antiangiogenic properties of apoA1‐mimic peptides in tumors have been reported[9]. We speculate that high apoA1 levels could protect retinal vascular endothelial cells to suppress retinal neovascularization and then prevent PDR development.
Pathological angiogenesis is mainly mediated by the vascular endothelial growth factor (VEGF) family[10], and VEGF‐A is a primary angiogenic growth factor. Anti‐VEGF therapy has been widely used clinically to treat retinal neovascularization.It is also known that 30%‐50% of DR patients are non‐responders or poor responders to VEGF inhibitor treatment,and repeated injections might also be associated with severe adverse effects[11‐12]. These clinical observations suggest that factors other than VEGF‐A may also play an important role in retinal neovascularization. The VEGF family consists of VEGF‐A, VEGF‐B, VEGF‐C, VEGF‐D, VEGF‐E, VEGF‐F,and placental growth factor (PlGF)[13], and the role of PlGF in ocular angiogenesis is less well understood than that of VEGF‐A[14]. The possible contribution of PlGF to diabetic retinopathy (DR) was already demonstrated in some studies,showing increased retinal, vitreous and aqueous PlGF levels in DR patients[15‐17]. This evidence shows that PlGF plays an essential role in DR. Moreover, PlGF is selectively associated with pathological angiogenesis, but not with physiological angiogenesis[18], with pathogenic pathways that appear distinct from those of VEGF‐A.In this study, we aimed to verify the inhibitory effect of apoA1 on retinal neovascularizationin vitroto explore the possible mechanism.
MATERIALS AND METHODS
Cell Culture and TreatmentHuman retinal vascular endothelial cells (HRECs) were purchased from Angioproteomie (Boston,MA, USA). The cells were maintained in endothelial cell medium (ECM; ScienCell, San Diego, California, USA)supplemented with 5% fetal bovine serum (FBS; ScienCell),1% penicillin/streptomycin (P/S, ScienCell), and 1%endothelial cell growth factor (ECGS; ScienCell). All cells were maintained in a humidified atmosphere of 5% CO2at 37℃. For hypoxia treatment groups, cells were put into hypoxia chambers (StemCell, Vancouver, Canada), which were infused with a gas mixture containing 1% O2, 5% CO2and 95% N2, and then placed in the incubator as described above.As mentioned above, the cells were incubated with endothelial cell medium (ECM; ScienCell, San Diego, California, USA)supplemented with 5% FBS before treatments, and then the medium was changed to ECM containing 1% FBS 24h prior to the experiment. For the wound healing assays, serum‐free ECM was used. To assess the antiangiogenic effects of apoA1 on hypoxia‐induced HRECs, the following groups were assigned according to oxygen concentration in the incubator and the lentivirus used: normal control group (21% O2,Ctrl);hypoxia group (1% O2, HY); empty vector control group (1%O2, EC); apoA1 overexpression group (1% O2, OP); U0126 group, pretreated with 15 μmol/L mitogen‐activated protein kinase kinase (MEK) inhibitor U0126 (T6223; Target Mol,Wellesley Hills, MA, USA) 1h before the hypoxia treatment.U0126 was dissolved in dimethyl sulfoxide (DMSO; Sigma‐Aldrich, St. Louis, MO, USA). The same volume of DMSO was used as a control. The HRECs and their media were collected 48h after hypoxia treatment for analysis.
Apolipoprotein A1 Gene OverexpressionThe human apoA1 overexpression lentiviral vector and its negative control vector were purchased from OBiO Technology (Shanghai,China). Lentivirus was produced in 293FT packaging cells and collected 48‐72h post‐infection. HRECs were seeded in 6‐well plates in ECM containing 5% FBS without antibiotics one day before transfection, such that they were 30%‐40% confluent at the time of transfection. HRECs were transduced with lentiviral particles [multiplicity of infection (MOI)=10] in the presence of 5 μg/mL polybrene for 12h. Then, fresh medium was replaced. Stably transfected cells were screened with 2.0 μg/mL puromycin (Invitrogen, Carlsbad, CA, USA) at 48h post‐infection. To maintain uniform conditions, all experiments were carried out by using passage 4‐7 HRECs.
ELISA for Vascular Endothelial Growth Factor and Placental Growth FactorCell culture media were collected after 48h of hypoxia or normoxia treatment. The concentrations of VEGF and PlGF in cell culture medium in different groups were measured by enzyme‐linked immunosorbent assay kits (ELISA; BD Biosciences, San Jose, CA, USA) with antibodies specific for human VEGF and PlGF according to the manufacturer’s instructions. Enzyme activity was measured with a Multi‐Mode Microplate Reader (Bio‐Tek, Winooski,VT, USA).
Scratch-wound AssayHRECs were seeded in 6‐well plates and incubated under different conditions as mentioned. After culturing for 48h, the cell density of each well reached 90%confluency, and then the monolayer cells were scratched gently using a 100 μL yellow micropipette tip. The scratched wells were then washed with phosphate buffer saline (PBS)three times, serum‐free medium was added, and the cells were exposed to hypoxic or normoxic environments. The wounds were photographed at 0 and 12h. The migration area was measured using Image J software 1.8.0 (National Institutes of Health, Bethesda, MD, USA). The migrated area was quantified relative to the area at 0, which was considered 100%.
Tube Formation Assay Briefly, a total of 150 μL of Matrigel(Corning, NY, USA) was added to a 48‐well plate and incubated at 37℃ for 30min to solidify the Matrigel. HRECs that had been cultured under appropriate conditions for 48h were seeded on solidified Matrigel at a density of 2×104cells/well. After 6h of incubation, images were captured by an inverted microscope(Zeiss, Thornwood, NY, USA). The total segment length formed by cells in Matrigel was counted directly from at least three random microscope fields.
Western Blot AnalysisTotal proteins were extracted from the HRECs using cell lysis buffer (#9803; Cell Signaling, Danvers,MA, USA) and quantified using a BCA protein analysis kit(#23227; Thermo, Carlsbad, CA, USA). An equal amount of protein (20 μg) was electrophoresed on a 10% denaturing polyacrylamide gel and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA, USA),which was then blocked with TBS/Tween‐20 containing 5%non‐fat milk for 1h at room temperature. The membranes were then incubated overnight at 4℃ with the following corresponding primary antibody diluents: anti‐pERK 1:1000(#9102; Cell Signaling, Danvers, MA, USA), anti‐p‐pERK 1:1000 (#9101S; CST), and anti‐β‐actin 1:1000 (#4970; CST).Then, the appropriate secondary antibodies were used. Target proteins on the membrane were visualized using an enhanced chemiluminescence (ECL) detection system (WBKLS0100;Millipore, Bedford, MA, USA). The band intensity was analyzed using Bio‐Rad Image Lab software (Bio‐Rad Laboratories, Hercules, CA, USA). At least three independent experiments were carried out.
Statistical AnalysisAll experiments were repeated at least three times, and results were represented as the mean±standard deviation (SD). One‐way ANOVA was used to evaluate the variations among groups, followed by Bonferroni’s multiple comparison test, which was used for comparison between two groups. All data were analyzed using GraphPad Prism 8.0.1 software for Windows. Differences were considered statistically significant atP<0.05.
RESULTS
Decreased of PlGF Level by Overexpressed apoA1 in Hypoxia-induced HRECsCells in the HY group and EC group were incubated under hypoxic conditions (1%) for 48h. Untransfected HRECs were cultured under normoxic conditions (21%) as the Ctrl group. Forty‐eight hours after incubation, the levels of free PlGF and VEGF in the media of each group were measured by ELISA. As shown in Figure 1,hypoxia‐induced HRECs secreted more PlGF than did HRECs under normoxic conditions (1.89±0.20‐fold,P=0.0003), and compared with the EC group, apoA1 overexpression significantly inhibited hypoxia‐induced PlGF expression (0.67±0.1‐fold,P=0.007). Similarly, the level of VEGF in the hypoxia group was significantly increased (2.71±0.23‐fold,P=0.0006).However, our data showed no significant differences in VEGF levels between the EC group and OP group.
Inhibition of Overexpressed apoA1 on Migration of Hypoxia-induced HRECTo confirm whether apoA1 affects the angiogenic process, we studied its effects on the migration ability of HRECs. Cells of each group were incubated under the indicated conditions. As wound‐healing experiments showed (Figure 2A), hypoxia obviously increased the migration ability (present as a larger migration area) of HRECs (2.21±0.27‐fold,P<0.0001) compared with the control condition. However, overexpression of apoA1 remarkably attenuated the migration (0.32±0.11‐fold,P<0.0001) induced by hypoxia compared with the empty vector (Figure 2B).This study showed that apoA1 could effectively inhibit the migration of HRECs induced by hypoxia.
Inhibition of Overexpressed apoA1 on Tube Formation of Hypoxia-induced HRECTo assess the functional role of apoA1 in angiogenesisin vitro, a tube formation assay was performed, which can mimic certain stages of angiogenesis.As shown (Figure 3A), the HRECs spontaneously formed capillary‐like structures after 6h of incubation on Matrigel.HRECs formed more capillary‐like structures (1.35±0.03‐fold,P<0.0001) under hypoxia than under the control conditions(Ctrl). However, compared with the empty vector, the overexpression of apoA1 obviously inhibited the increase in capillary‐like structures (0.66±0.01‐fold,P<0.0001) induced by hypoxia (Figure 3B). This study indicated that apoA1 effectively inhibited the formation of capillary‐like structures in HRECs induced by hypoxia.
Effects of ApoA1 Overexpression on MEK/ERK Signaling Pathways in Hypoxia-induced HRECsCells with different apoA1 expression levels were used: OP group and EC group were incubated under hypoxic conditions for 48h.Untransfected HRECs were used as the control group; the effects of apoA1 on signaling pathways were evaluated by Western blotting. As shown (Figure 4B), the group with apoA1 overexpression showed markedly decreased p‐ERK levels(0.6±0.11‐fold,P=0.025) compared with the EC group, while the levels of total ERK in both groups were not different.Collectively, these results suggested that apoA1 inhibited hypoxia‐induced PlGF secretion, migration and tube formation in HRECs and might be related to the dephosphorylation of ERK1/2.
MEK/ERK Signaling Pathways Mediate PlGF Secretion,Migration, and Tube Formation in Hypoxia-induced HRECsHRECs from different groups were incubated under the indicated conditions and with the indicated inhibitors for 48h. ERK signaling was blocked with the MEK/ERK inhibitor U0126. The same volume of DMSO was used as a control.Forty‐eight hours after incubation, the levels of free PlGF and VEGF in the media of each group were measured by ELISA.As shown (Figure 4C), hypoxia‐induced HRECs secreted more PlGF (1.93±0.19‐fold,P<0.0001) than HRECs under normoxic conditions. However, the OP group showed significantly inhibited hypoxia‐induced PlGF expression compared with the EC group (0.68±0.11‐fold,P=0.0051). Furthermore,pretreatment with U0126 further enhanced the inhibitory effect of apoA1 (0.4±0.06‐fold,P=0.0004). A consistent phenomenon was also observed in the wound‐healing experiment and tube formation assay (Figures 2, 3).Therefore, blocking the MEK/ERK signaling pathway could reduce hypoxia‐induced PlGF secretion in HRECs, as could angiogenesis in HRECs. From the above results, a conclusion can be drawn that apoA1 might suppressed ERK pathways and modulated the expression of PlGF.
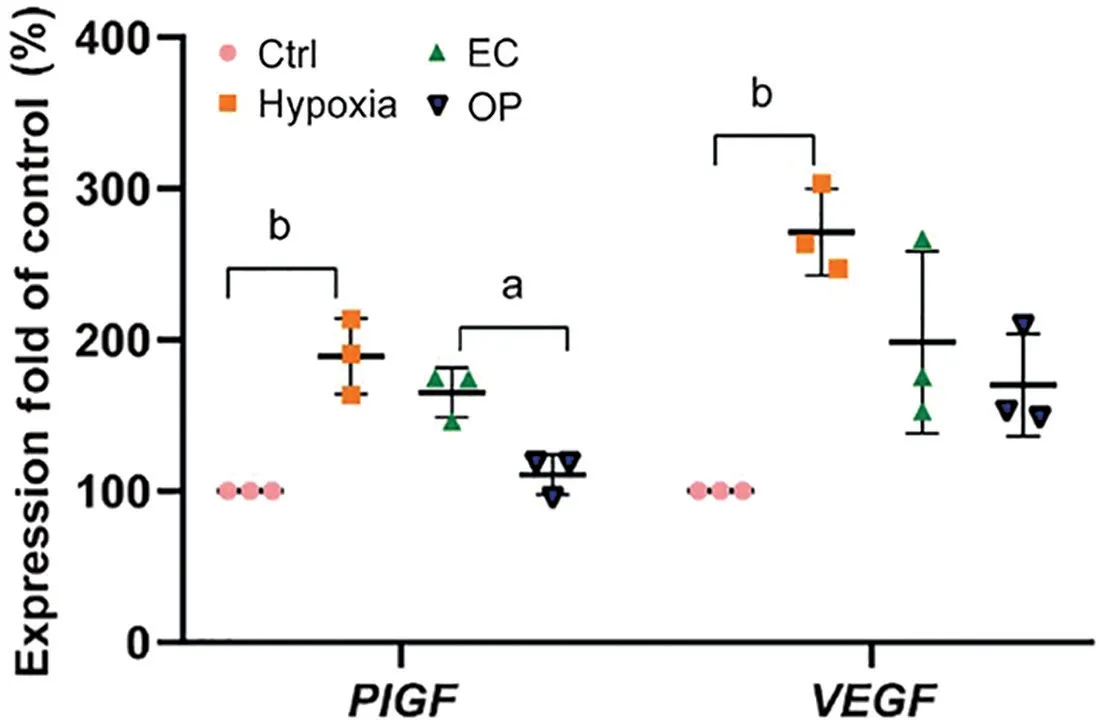
Figure 1 Effect of apoA1 on PlGF and VEGF in hypoxia-induced HRECs Ctrl: Normal control group; EC: Empty vector control group;OP: ApoA1 overexpression group; VEGF: Vascular endothelial growth factor; PlGF: Placental growth factor; HRECs: Human retinal vascular endothelial cells. aP<0.05, bP<0.002.

Figure 2 Effects of apoA1 on the migration ability of hypoxiainduced HRECs Ctrl: Normal control group; EC: Empty vector control group; OP: ApoA1 overexpression group; HRECs: Human retinal vascular endothelial cells. cP<0.0001.

Figure 3 Effects of apoA1 on the tube formation ability of hypoxiainduced HRECs Ctrl: Normal control group; EC: Empty vector control group; OP: ApoA1 overexpression group; HRECs: Human retinal vascular endothelial cells. aP<0.05, cP<0.0001.

Figure 4 Effect of apoA1 on the phosphorylation of ERK1/2 in hypoxia-induced HRECs and the effect of a MEK inhibitor (U0126)on hypoxia-induced PlGF Ctrl: Normal control group; EC: Empty vector control group; OP: ApoA1 overexpression group; PlGF:Placental growth factor; HRECs: Human retinal vascular endothelial cells; MEK: Mitogen-activated protein kinase kinase. aP<0.05,bP<0.002, cP<0.0001, ns: Not significant.
DISCUSSION
Retinal neovascularization is the essential pathological basis of many blinding eye diseases, including PDR. As mentioned, VEGF and PlGF are two major growth factors involved in this pathological process. In this study, we showed that overexpression of apoA1 inhibited angiogenesis and suppressed PlGF expression in hypoxia‐induced HRECs.Therefore, apoA1 had notable antiangiogenic effects on HRECs.
We proposed the scientific hypothesis that high apoA1 levels might suppress retinal neovascularization based on previous clinical observations and further verified the antiangiogenic effect of apoA1 on hypoxia‐induced HRECs for the first time.Moreover, previous studies have mostly focused on VEGF rather than PlGF, while we have paid attention to the important role of PlGF, which has functions that are distinct from those of VEGF‐A in the pathogenesis of hypoxia‐induced HRECs.Surprisingly, we found that apoA1 decreased the level of PlGF in hypoxia‐induced HRECs.
There is only a small amount of ophthalmology research on apoA1, mostly focusing on three diseases: DR, age‐related maculopathy (AMD) and rhegmatogenous retinal detachment(RRD). The presence of apoA1 in the subretinal fluid (SRF)of RRD patients showed that only low molecular size plasma proteins escape more readily from the choriocapillaris‐pigment epithelium complex[19]. The relationship between the apoA1 mimetic peptide L‐4F and AMD has been studied.L‐4F is a potent modifier of plasma membranes and has anti‐inflammatory and antiatherogenic functions. Rudolfet al[20]demonstrated a highly effective pharmacological reduction in esterified cholesterol and restoration of Bruch’s membrane ultrastructure by intravitreally injecting 4F in a murine model. Then, the next year, the same authors delivered L‐4F intravitreally and induced a substantial pharmacologic reduction in Bruch’s membrane lipids and the restoration of ultrastructure in aged nonhuman primates[21]. All these data support L‐4F as a promising new candidate for treating the underlying cause of AMD. Many recent studies have explored the relationship between apoA1 levels and the role of lipids in the pathogenesis of DR. The serum apoA1 level was lower in a PDR group than in a nonproliferative diabetic retinopathy(NPDR) group[22], and serum apoA1 was statistically significantly associated with a reduced likelihood of having more severe DR levels in diabetic patients[4,23]. Simóet al[24]documented a higher level of apoA1 in the vitreous humor of DR patients. Interestingly, the same authors also found apoA1 overexpression in the retina of diabetic donors and hypothesized that the increased production of retinal apoA1 was a protective compensatory mechanism against DR due to its function of transport of lipids within the retina[25].
ApoA1 and its mimic peptides are reported to have anti‐inflammatory and antioxidant functions and to be involved in the intraretinal reverse transport of lipids[6‐8]. As demonstrated,apoA1‐mimic peptides inhibited neovascularization in mouse ovarian cancer models, which was associated with the inhibited phosphorylation of ERK and AKT[9]. The ERK signaling pathway has been recognized to mediate a wide range of functions, including proliferation, growth, and survival[26]. In the current study, we found that the overexpression of apoA1 in HRECs inhibited the protein levels of phosphorylated ERK1/2.Moreover, blocking the MEK/ERK pathway had similar effects as overexpressing apoA1, which led to a decrease in PlGF secretion. Based on these findings, we proposed that the antiangiogenic effect of apoA1 was achieved at least in part by regulating the ERK pathway.
Our outcomes highlighted the important role of PlGF in the pathogenesis of hypoxia‐induced HRECs. PlGF expression was regulated by hypoxia[27], and PlGF enhanced pathological angiogenesis in endothelial cells[28], which was consistent with our outcomes. PlGF is different from VEGF‐A in the following aspects. First, PlGF‐deficient mice had normal vascular development, suggesting that PlGF does not affect physiological angiogenesis[29‐30]. Furthermore, we analyzed the morphology of quiescent vessels in different healthy organs and found that treatment with anti‐VEGFR2, but not anti‐PlGF, reduced the number of capillary branches in the trachea and capillary profiles in the thyroid gland. Moreover, the addition of anti‐PlGF did not aggravate the adverse effect of anti‐VEGFR2 on vessel pruning[30]. There was also a clinical study describing that anti‐VEGF treatment in DR patients was associated with the thinning of the retinal nerve fiber layer and with retinal ganglion cell apoptosis[31]. PlGF regulates a range of vascular, neural and glial cell responses that are different from VEGF‐A. Studies have shown that PlGF deletion or inhibition could reduce neovascularization, retinal leakage,inflammation and gliosis without affecting healthy vasculature or inducing neuronal degeneration[32‐34]. These experimental results were recently confirmed in a preclinical study showing that the intravitreal injection of anti‐PlGF antibody was equally effective as aflibercept (recombinant fusion protein inhibiting both VEGF and PlGF) in reducing the choroidal neovascularization‐localized inflammatory response, whereas treatments with anti‐VEGFR2 or anti‐VEGF had no effect.Moreover, anti‐PlGF antibody showed efficacy comparable to anti‐VEGF against vascular leakage without triggering a neurodegenerative response[33]. In summary, PIGF might be a potential therapeutic target in ocular diseases, especially retinal diseases.
In summary, apoA1 inhibits angiogenesis at least in part by suppressing ERK1/2 and suppresses PlGF expression in hypoxia‐induced HRECs. In addition, PlGF was selectively associated with pathological angiogenesis but not physiological angiogenesis. These findings suggest that apoA1 may be a promising therapeutic for treating retinal neovascularization diseases.
ACKNOWLEDGEMENTS
Foundations:Supported by the National Natural Science Foundation of China (No.81500735; No.81970807).
Conflicts of Interest: Hu J, None;Chen ZT,None;Su KY,None;Lian Y,None;Lu L,None;Hu ADN, None.
 International Journal of Ophthalmology2023年1期
International Journal of Ophthalmology2023年1期
- International Journal of Ophthalmology的其它文章
- Instructions for Authors
- Morphological and functional changes in the macular area in diabetic macular edema after a single intravitreal injection of aflibercept
- Macular vascularisation changes analysed using OCT angiography after successful rhegmatogenous retinal detachment repair
- Comparison of success rate and intraocular pressure spikes between selective laser trabeculoplasty and micropulse laser trabeculoplasty in African American and Hispanic patients
- Efficacy of custom-made soft keratoconus lenses on corneal aberrations and photic phenomena in patients with keratoconus: a corneal topography imaging based study
- Clinical observation of recombinant human nerve growth factor in the treatment of neurotrophic keratitis