
临沂市一例Omicron BA.5.2变异株全基因组序列分析
2023-01-29 13:25季圣翔刘祥亮
山东医学高等专科学校学报 2022年6期
张 林,季圣翔,刘祥亮
(临沂市疾病预防控制中心,山东 临沂 276000)
自新冠病毒疫情爆发以来,已经多次出现变异。2022年11月南非发现的Omicron变异株相比其他亚型具有更高的传播性、更高的病毒结合亲和力和更高的抗体逃逸等特点,在全球迅速蔓延,成为新冠病毒的主要流行株[1]。Omicron BA.5.2变异株在国内外引起大量疫情暴发,因此对Omicron BA.5.2全基因组分析及溯源对新冠疫情控制具有重要意义。
1 材料与方法
1.1材料 本研究的病毒株BA.5.2来源于2022年10月14日—2022年11月10日临沂市Omicron变异株引起的新冠病毒感染者。
1.2方法
1.2.1流行病学分析 根据新型冠状病毒防控方案(第九版)进行流行病学调查,根据调查结果进行分析。
1.2.2SARS-CoV-2核酸检测 使用艾克韦核酸提取试剂盒(山东艾克韦,批号:2022040201)提取病毒基因组RNA。使用新型冠状病毒荧光定量PCR试剂盒(武汉明德,批号:220818)进行检测。
1.2.3全基因组测序 使用新型冠状病毒全基因组低载量捕获试剂盒(微未来,中国北京,批号:2022OMP2/2-075)对SARS-CoV-2基因组RNA进行逆转录和特异性扩增。使用Nextera XT Library Prep Kit(因美纳,美国,批号:20634442)构建Illunima文库并利用MiSeq(因美纳,美国,批号:20637219)进行二代测序。
1.3数据处理 使用CLC Genomics workbench 22 Properties软件,以武汉株NC 045512作为参考序列,对下机数据进行拼接和一致化分析。从GISAID筛选并下载亚洲地区上传GISAID的Omicron BA.5.2变异株的序列132条。使用CLC Genomics workbench 22 Properties中的建树分析工具,对8条全基因组核苷酸序列和132条Omicron BA.5.2变异株的序列进行比对,并基于最大似然法构建系统进化树,Bootstrap值设置为1000,以评估可靠性。运用突变位点和缺失等信息的在线分析工具“Nextclade”分析新冠基因序列变异位点。
2 结果
2.1流行病学特征 本次疫情流调结果指向10月9日—12日自驾途径新疆、内蒙等地的外地病例Case1。Case2的职业是三轮车跑腿,10月14日Case1的同时空密接;Case3~5为10月14日早6时20分同Case1一起就餐者;Case6、7为10月17日同Case3一起做核酸检测且同在20合1管中;Case8为Case3的同事;Case9为Case5的姐姐,10月15日两人同住;Case10为Case2的妻子;Case11为Case2、10的重孙女。感染者的流调关系见封三图1。
2.2SARS-CoV-2核酸检测结果 对36名涉疫人员进行核酸检测,其中12份鼻咽拭子样本的检测结果为阳性,ORF基因和N基因的中位数M(P25,P75)分别为27.05(21.06,34.95)和28.29(22.23,35.11)。潜伏期中位数M(P25,P75)为2.5 d(1.25,3.75)。见表1。
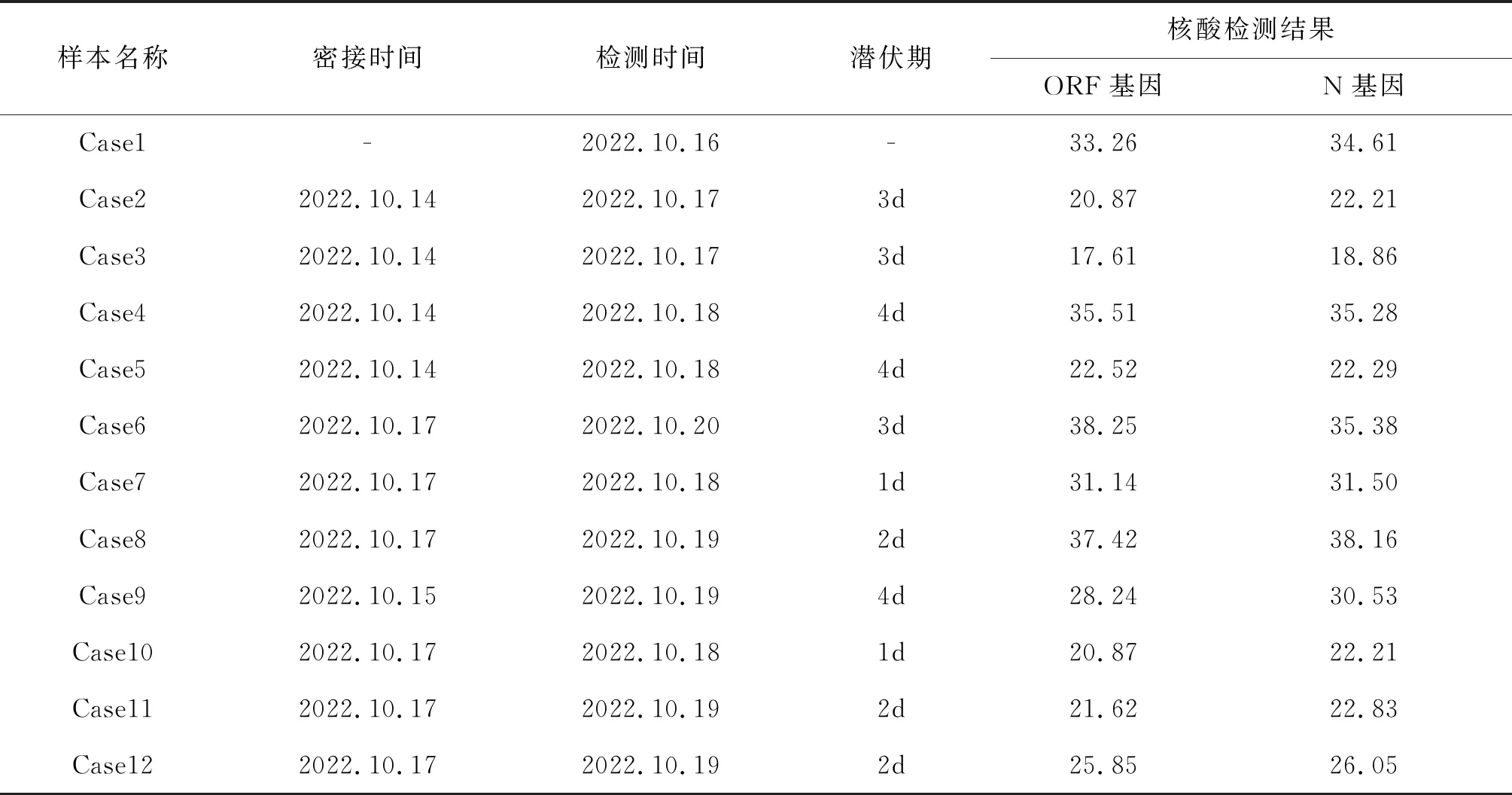
表1 SARS-CoV-2核酸检测阳性汇总表
2.3全基因组测序结果 12例感染者的样本行全基因组测序,得到8条全基因组序列分别为Case2~4、Case7、Case9~12。Case1、Case5、Case6、Case8因CT值较高,病毒载量低未获得全基因组序列。从进化树上可以发现,Case2~4、Case7、Case9~12以及印度尼西亚检出编号为15783704、马来西亚检出编号为15535797、15535803、1553579、印度检出编号为15422884的样本基因序列聚成一簇,亲缘关系同其他序列相比较近。同其他序列不能聚成一簇,亲缘关系相对较远。Case10、Case11较其他序列亲缘关系更近。见封三图2。
2.4变异位点分析 “Nextclade”分析结果显示,Case2~4、Case7、Case9、Case12测序结果为奥密克戎BA.5.2分支,共享74核苷酸变异位点。Case10、Case11的基因序列完全一致,较Case2等人新增T7897C核苷酸变异位点。T7897C核苷酸变异位点位于ORF1a基因,该突变不引起氨基酸的变化。同GISAID上下载序列相比,此次疫情爆发中测定的全基因组序列新增C884T、C7728T、C18647T、G23608T四个核苷酸变异位点。其中C884T、C7728T核苷酸变异位点引起ORF1a基因中R702C、S2488F两处氨基酸发生改变;C18647T引起ORF1b中P1727L氨基酸改变;G23608T引起S蛋白N501Y氨基酸改变。见表2。

表2 全基因组序列变异分析表
3 讨论
本研究中Omicron BA.5.2变异株潜伏期最长为4 d,中位数中位数M为2.5 d。疫情发生后5 d内就出现了4代病例,与其他研究结果比较潜伏期和疫情传播代际明显缩短[2-4]。可以推测出Omicron BA.5.2变异株的传播能力增强,但是隐匿性较之前的毒株有所减弱。虽然因CT值较高,未能获得Case1的全基因组序列,但是获得了关键病例Case2和Case3的全基因组序列。通过构建系统进化树和“Nextclade”分析发现,Case2、Case3、Case4的全基因组序列完全一致,说明三者属于同一传播链条。Case10、Case11全基因组序列完全一致,较其他6份样本新增T7897C核苷酸变异位点,说明病毒是由Case2传至Case10再由Case10传给Case11。结合流行病学调查和感染者的核酸检测阳性的时间,可以推测出此次疫情是由省外来人Case1输入引起的暴发。
此次Omicron BA.5.2变异株同亚洲其他国家的Omicron BA.5.2变异株新增C884T、C7728T、C18647T、G23608T四个核苷酸变异位点。这4个变异位点引起ORF1ab中的三个氨基酸改变和S蛋白中N501Y氨基酸的改变,S蛋白的变化可能导致新冠毒株传播性和抗体逃逸能力发生变化[5]。此次疫情Omicron BA.5.2变异株的潜伏期和疫情传播代际的显著性缩短可能同S蛋白中N501Y氨基酸的改变相关。
流行病学调查显示,Case2的传播途径为同时空交集,Case6、Case7的感染原因为核酸检测过程中个人防护不到位。但是36名涉疫人员中有24人在有效个人防护的情况下并未造成感染,说明做好个人防护的重要性。此次疫情持续5 d后未在同一传播链条上在发现新的感染者,说明应对本次疫情采取的防疫措施是行之有效的。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
中国慈善家(2022年3期)2022-06-14
现代苏州(2022年9期)2022-05-26
快乐语文(2021年34期)2022-01-18
中国(俄文)(2020年8期)2020-11-23
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
中国饲料(2019年19期)2019-03-25
