
贝母属植物叶绿体基因组结构与系统发育的关系
2023-01-12 06:03:12马晓君郭宗雷王信宏玄静田静刘欢隋园园束靖
山东农业大学学报(自然科学版) 2022年5期
马晓君,郭宗雷,王信宏,玄静,田静,刘欢,隋园园,束靖
贝母属植物叶绿体基因组结构与系统发育的关系
马晓君,郭宗雷,王信宏,玄静,田静,刘欢,隋园园,束靖*
山东农业工程学院, 山东 济南 250000
贝母属植物形态相似,难以区分。通过对叶绿体基因组进行分析,可以为贝母属植物的系统发育、分子鉴定和开发利用提供理论依据。我们基于已发表的贝母属物种的叶绿体基因组序列,利用相关生物信息学方法对其叶绿体基因组进行分析。结果显示,贝母属29个物种叶绿体基因组大小在151058~152434 bp。基因数量为128~133个,rRNA编码基因数目稳定,蛋白质编码基因和tRNA编码基因存在差异。四分区分析发现,IRb/SSC边界存在明显差异。串联重复和单核苷酸重复含量最丰富。共线性分析未检测到基因重排,其叶绿体基因组高度保守。基于叶绿体基因组的系统发育分析表明,贝母属植物分为4个主要分支。对贝母叶绿体基因组进行核苷酸多态性分析,发现IR比单拷贝区更保守,非编码区的突变频率比编码区高。
贝母属植物; 叶绿体; 基因; 系统发育
中药材“贝母”属百合科(Liliaceae)贝母属(),绝大多数是多年生草本植物。入药部分为的鳞茎,具有清热润肺、化痰止咳等功效[1]。近年来,多位学者已经对国产贝母属的植物进行了系统分类研究,发表了许多新种和新变种,使国产贝母属植物“种”数已达80个,“变种”名称数已达52个[2]。由于贝母属植物在形态上十分相近,难以在形态上进行区分,在中药材市场中常出现以次充好、以伪充真等问题[3]。
被子植物叶绿体通常具有由一对倒置重复(inverted repeat, IR)区,小单拷贝(small single-copy, SSC)区,大单拷贝(large single-copy, LSC)区组成的四分体结构。众多研究表明,叶绿体基因组结构和序列保守,在分子标记开发、遗传育种、物种鉴定及系统进化研究等方面具有重要作用[4, 5]。尤其是随着DNA测序技术的快速发展,越来越多地物种完成叶绿体基因组测序[6]。并且叶绿体基因组相较于DNA条形码具有更高的分辨率,已成为中草药鉴定、分类的重要工具[7]。本试验基于已发表的贝母属叶绿体基因组序列,利用生物信息学方法对其叶绿体基因组的特征、基因数量与长度、结构变异、重复序列、基因重排、系统发育和序列多态性进行分析,为贝母属植物的系统发育、分子鉴定和开发利用提供理论依据。
1 数据收集和研究方法
1.1 数据收集
在Genbank数据库(https://www.ncbi.nlm.nih.gov/genome/organelle/)下载已发表的29个百合科贝母属植物的叶绿体基因组序列信息,收集其物种名称、GenBank登录号等(见表1)。

表1 29种贝母属植物叶绿体基因组注释信息
1.2 叶绿体基因组注释
利用在线网站CPGAVAS2(http://47.96.249.172:16019/analyzer/home)进行叶绿体基因组的注释[8]。Excel统计叶绿体基因组长度、基因数量、四分区长度等信息。
1.3 叶绿体基因组比较
通过IRSCOPE(https://irscope.shinyapps.io/irapp/)对叶绿体基因组四分区进行可视化[9]。以叶绿体基因组作为参考序列,通过在线网站mVISTA(http://genome.lbl.gov/vista/mvista/submit.shtml)中的Shuffle-Lagan模式进行全基因组比较[10];利用Mauve软件对叶绿体基因组进行全局比对,并进行共线性分析[11]。
1.4 SSR与重复序列分析
通过在线分析网站REPuter(https://bibiserv.cebitec.uni-bielefeld.de/reputer/)查找散在重复序列和回文重复序列[12],参数设置为:汉明距离3,最大计算重复次数90,最小重复单元长度30。串联序列通过TRF(Tandem Repeats Finder)(http://tandem.bu.edu/trf/trf.html)查找[13],使用默认参数。简单重复序列(Simple Sequence Repeats, SSRs)通过MISA网站(https://webblast.ipk-gatersleben.de/misa/)查找[14],重复次数阈值设置为:单核苷酸10、二核苷酸5、三核苷酸4、四核苷酸3、五核苷酸3、六核苷酸3。
2.5 构建系统发育树
本研究中29个物种的叶绿体基因组长度和四分区长度差异不大,选择叶绿体全基因组数据进行分析,通过最大似然法(maximum likelihood, ML)构建系统发育树。使用百合科百合属的、、作为外类群[15],与贝母属一起构建系统发育树。使用MAFFT软件对叶绿体基因组进行多序列比对[16],通过MEGA获得最佳替代模型并构建ML系统发育树[17],bootstrap次数为1000次。
2.6 序列多态性分析
使用MAFFT进行多序列比对[16],将比对结果导入DNAsp[18],计算叶绿体基因组序列的核苷酸多态性(pi),参数设置为:window length 600,step size 200。
2 结果与分析
2.1 叶绿体基因组结构特征
通过叶绿体基因组注释信息(见表1)发现,本研究中29个物种的叶绿体基因组总长度差距不大,最长的长度为152434 bp,最短的长度为151058 bp,相差约1.4 kb。叶绿体基因组包括一对反向重复区(IR)、大单拷贝区(LSC)和小单拷贝区(SSC)。本研究中LSC区长度在81339~82130 bp之间,最长的为,最短的是,相差791 bp;SSC区长度在16962~17684 bp之间,最长的为,最短的为,相差722 bp;两个IR区域等长,在26123~26574 bp之间,其中长度最长,长度最短,相差451 bp。
2.2 基因数量
29个百合科贝母属植物的叶绿体基因总数在128~133之间(表1),其中17个物种的基因数量为129,占大多数。rRNA编码基因数目稳定,全部为8个;tRAN数大多数为38,只有3个物种含有37个tRNA;蛋白质编码基因数量在82~87之间,其中14个物种含有83个(见表1),可见基因总数差异主要是由蛋白编码基因差异引起的。
2.3 四分区边界分析
叶绿体四分区边界为LSC/IRb、IRb/SSC、SSC/IRa、IRa/LSC。通过分析各边界叶绿体基因的分布状况,可以分析贝母叶绿体在进化过程中IR区收缩与扩张现象。如图1可以得知,LSC/IRb边界位于和基因内,由于IR区域的收缩和扩张,有5个物种的全部位于LSC区域;中的在边界几乎均匀分布;其余23个物种的有236~268 bp位于LSC区域,11~43 bp进入IRb区域。IRb/SSC边界处、基因分布存在较大差异,有16个物种的全部位于IRb区内,其中的长度更短;12个物种的大部分位于IRb区,16~121 bp进入SSC区域;的基因丢失;大多数物种位于SSC区内,5个物种的跨过IRb/SSC边界,有8~33 bp位于IRb区。SSC/IRa边界处长度与分布大致相似,均为4245~4394 bp位于SSC区,1147~1263 bp位于IRa区;但和的长度较短且位于SSC区的长度发生变化。IRa/LSC边界位于和之间,只有的IRa/LSC边界位于基因内。

图1 29个贝母属植物叶绿体基因组四分区边界比较
2.4 重复序列及SSR分析
在贝母属29个物种中共发现1853个重复序列,包括28.87%散在重复、33.51%回文重复和37.61%串联重复(图2A,B)。单个物种的重复序列数目大多在57~67之间,但仅含有51个,4个物种的重复序列数超过70个。SSR共发现2176个。其中单核苷酸数目最多,占67.69%(1473个),二核苷酸占17.88%(389个),三核苷酸占3.08%(67个),四核苷酸占10.11%(220个),五核苷酸占1.19%(26个),仅有含有1个六核苷酸(图2C,D)。29个物种中,SSR总数在59~83之间,其中数目最少,数目最多。单核苷酸数为39~58;二核苷酸数为9~16;三核苷酸数为1~6;四核苷酸数为6~9;五核苷酸数目为0~3之间,其中9种贝母不含五核苷酸重复。

(A)3种重复类型的比例The ratio of three repeated types;(B)3种重复类型的数目The number of three repeats types;(C)不同类型SSR的比例The ratio of different SSR;(D)不同类型SSR的数目The number of different SSR
2.5 叶绿体基因组水平对比和共线性分析
全基因组比对结果表明(图3),整个叶绿体基因组高度保守,且IR比单拷贝区更保守,非编码区的突变频率比编码区高。、、基因的编码区比其他基因的编码区更具可变性。共线性分析结果显示没有检测到大片断基因重排现象(图4),也表明贝母属的29个物种相对保守。

图3 29种贝母属植物叶绿体基因组比对
注:轴代表叶绿体基因组中的坐标,轴表示对齐区域的序列相似性在50%~100%。箭头表示基因及转录方向。
Note: The x-axis represents the coordinate in the chloroplast genome. The y-axis indicates that the sequence similarity of the aligned region is between 50% and 100%. Arrows indicate genes and transcriptional direction.

图4 29种贝母属植物叶绿体基因组共线性分析
2.6 系统发育树分析
使用ML法对29种贝母属植物及3种百合科植物的叶绿体全基因组序列进行系统发育树分析,最佳替代模型为GTR+G+I。进化树各节点分支分辨率高(图5),位于进化树基部,属于较早分化的类群,且与百合属聚类为一支;是单独的一支,与其他物种为姐妹类群;、、、聚为一支;与聚为一支;、、、、聚为一支;其余16种贝母聚类为一支。
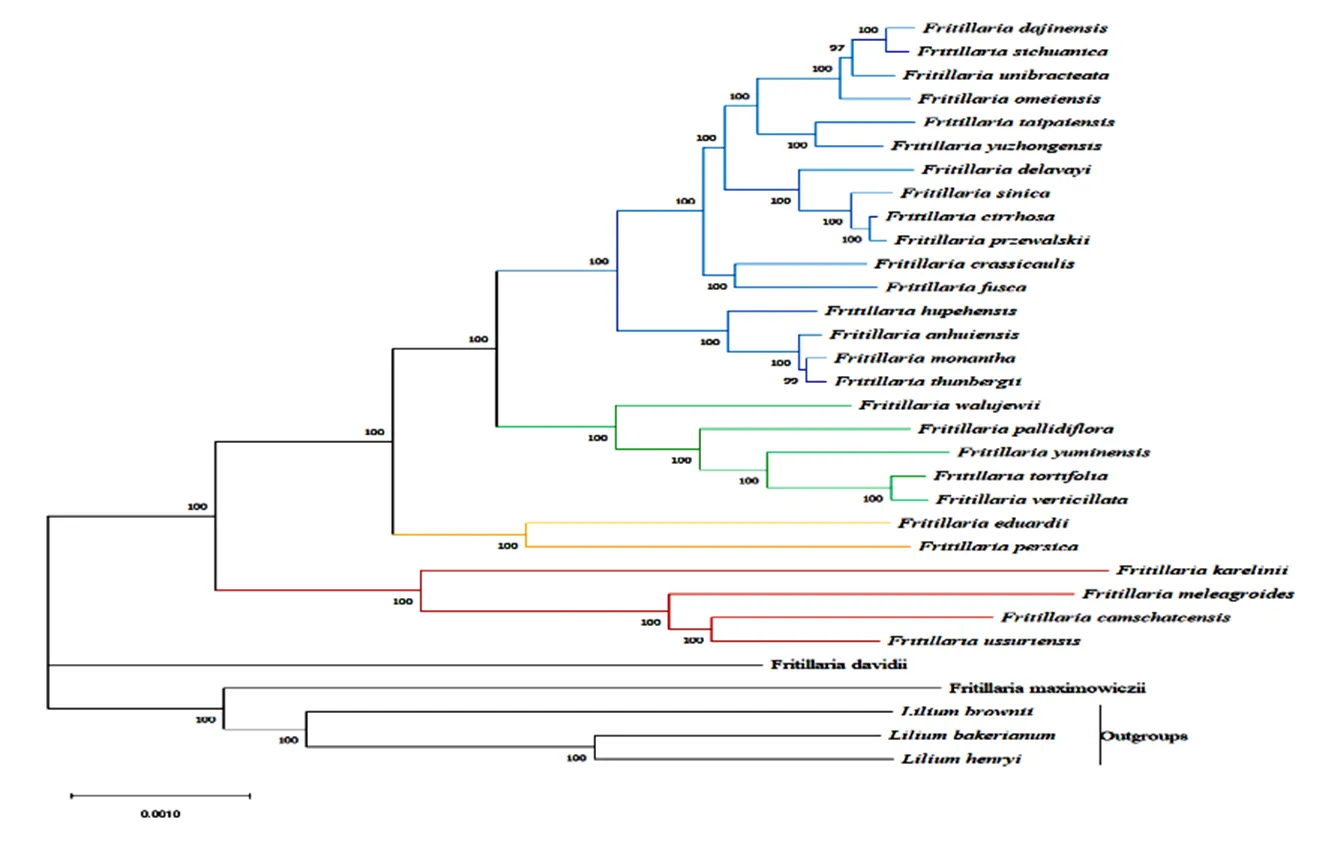
图5 基于叶绿体全基因组序列构建的ML树
2.7 序列多态性分析
共检测到718个核苷酸多态性位点,核苷酸多态性范围为0~0.02176(图6),平均值为0.005254248,-、-、-等基因或基因间隔区具有较高的变异,它们的核苷酸多态性均大于0.02,其中-的多态性最高,为0.02176,-和-的多态性分别为0.02106和0.02033。
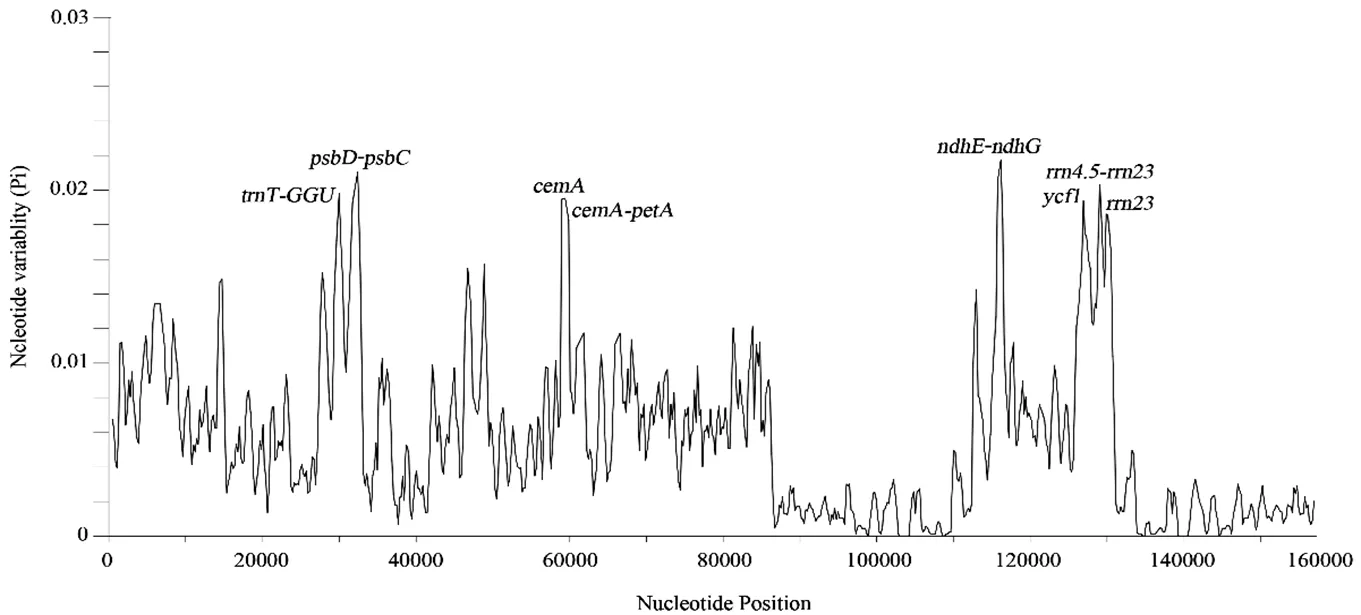
图6 29种贝母属植物叶绿体基因组滑动窗口分析
3 讨论与结论
贝母属29个物种叶绿体基因组的大小在152434~151058 bp之间,LSC区、SSC区、IR区长度与被子植物叶绿体基因组长度相吻合[19]。本研究中,各物种间基因组大小相差约1.4 kb左右,LSC、SSC以及IR区域相差分别为0.8 kb、0.7 kb以及0.45 kb左右。叶绿体基因组IR区的收缩与扩张普遍发生在被子植物进化过程中[20],是基因组结构变异的重要原因。贝母属叶绿体基因组结构、基因数量和顺序高度保守[21]。29种贝母属植物的4个边界区分别跨过、-、、-。但IRb/SSC边界处、的分布在不同物种中存在一定差异,贝母属叶绿体基因组长度变异可能是IR和SSC边界变化引起的[22]。与百合科植物类似,贝母属植物叶绿体基因组中的LSC/IRb边界大多扩展到基因,这可能是百合科贝母属植物的共同祖征[23,24]。
叶绿体共线性分析没有检测到基因重排现象,全基因组序列比对也显示叶绿体基因组序列相似度很高,其IR区比单拷贝区更保守,非编码区的突变频率比编码区高,与前人研究结果一致[21,25]。长重复序列与SSR在叶绿体基因组结构变异中具有重要作用,常用于药用植物物种鉴定、群体遗传学研究[26,27]。在29种贝母属植物叶绿体基因组中单核苷酸数目最多,因此单核苷酸可能比其他类型的SSR对贝母遗传变异的贡献更大。有研究表明、-、能更好地识别分辨贝母属植物[28],但在本研究中-、的pi值不高,的pi值为0.01938。也被认为是陆地植物最有可能的叶绿体DNA条形码[29],有望于开发成为高效鉴别贝母属植物DNA条形码。
本研究构建的构建ML树中,大部分系统发育关系与Zhang T、Chen Q等的结果相似[27,28],除与外,主要聚为4个主要分支,与百合属聚类为一支,并且分辨率达到100 BP而非Zhang T等的75 BP[27]。其余不同可能是因为所使用的叶绿体基因组数据不同导致。
[1] 徐顺连,曾中兰,林青,等.贝母化学成分及药理作用的研究[J].青海草业,2021,30(1):43-46
[2] 肖培根,姜艳,李萍,等.中药贝母的基原植物和药用亲缘学的研究[J].植物分类学报,2007(4):473-487
[3] 罗焜,马培,姚辉,等.基于ITS2序列鉴定川贝母及其混伪品基原植物[J].世界科学技术(中医药现代化),2012,14(1):1153-1158
[4] Li HT, Yi TS, Gao LM,. Origin of angiosperms and the puzzle of the Jurassic gap [J]. Nat Plants, 2019,5(5):461-70
[5] Nie Y, Foster CSP, Zhu T,Accounting for uncertainty in the evolutionary timescale of green plants through clock-partitioning and fossil calibration strategies [J]. Syst Biol, 2020,69(1):1-16
[6] Dobrogojski J, Adamiec M, Luciński R. The chloroplast genome: a review [J]. Acta Physiologiae Plantarum, 2020,42(6):98
[7] 林小涵,刘志华,李卿,等.药用植物叶绿体基因组研究[J].世界科学技术(中医药现代化),2010,12(3):442-446
[8] Shi L, Chen H, Jiang M,. CPGAVAS2, an integrated plastome sequence annotator and analyzer [J]. Nucleic Acids Res, 2019,47(1):65-73
[9] Amiryousefi A, Hyvonen J, Poczai P. IRscope: an online program to visualize the junction sites of chloroplast genomes [J]. Bioinformatics, 2018,34(17):3030-3031
[10] Frazer KA, Pachter L, Poliakov A,. VISTA: computational tools for comparative genomics [J]. Nucleic Acids Res, 2004,32:273-279
[11] Darling AC, Mau B, Blattner FR,. Mauve: multiple alignment of conserved genomic sequence with rearrangements [J]. Genome Res, 2004,14(7):1394-1403
[12] Kurtz S, Choudhuri JV, Ohlebusch E,. REPuter: the manifold applications of repeat analysis on a genomic scale [J]. Nucleic Acids Res, 2001,29(22):4633-4642
[13] Benson G. Tandem repeats finder: a program to analyze DNA sequences [J]. Nucleic Acids Res, 1999,27(2):573-580
[14] Beier S, Thiel T, Munch T,. MISA-web: a web server for microsatellite prediction [J]. Bioinformatics, 2017,33(16):2583-2585
[15] Li Y, Zhang Z, Lv G. The complete chloroplast genome of, a rare and endangered species endemic to China [J]. Mitochondrial DNA B Resour, 2017,2(2):913-914
[16] Katoh K, Misawa K, Kuma K,. MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform [J]. Nucleic Acids Res, 2002,30(14):3059-3066
[17] Tamura K, Stecher G, Kumar S. MEGA11: Molecular evolutionary genetics analysis version 11 [J]. Mol Biol Evol, 2021,38(7):3022-3027
[18] Rozas J, Ferrer-Mata A, Sanchez-Delbarrio JC,. DnaSP 6: DNA sequence polymorphism analysis of large data sets [J]. Mol Biol Evol, 2017,34(12):3299-3302
[19] Zhang T, Fang Y, Wang X,. The complete chloroplast and mitochondrial genome sequences of: insights into the evolution of plant organellar genomes [J]. PLoS One, 2012,7(1):e30531
[20] Fu CN, Li HT, Milne R,. Comparative analyses of plastid genomes from fourteen Cornales species: inferences for phylogenetic relationships and genome evolution [J]. BMC Genomics, 2017,18(1):956
[21] Huang J, Yu Y, Liu YM,. Comparative chloroplast genomics of(liliaceae), inferences for phylogenetic relationships betweenandand plastome evolution [J]. Plants, 2020,9(2):133-148
[22] Bi Y, Zhang MF, Xue J,. Chloroplast genomic resources for phylogeny and DNA barcoding: a case study on Fritillaria [J]. Sci Rep, 2018,8(1):1184
[23] Li P, Lu RS, Xu WQ,. Comparative genomics and phylogenomics of east asian tulips (Amana, Liliaceae) [J]. Front Plant Sci, 2017,8:451
[24] Li Q, Li Y, Song J,. High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy [J]. New Phytologist, 2014,204(4):1041-1049
[25] Park I, Kim WJ, Yeo SM,. The complete chloroplast genome sequences ofmaxim. andD. don, and comparative analysis with otherspecies [J]. Molecules, 2017,22(6):982
[26] 田星,刘莹莹,张颖敏,等.藜芦属药用植物的叶绿体基因组比较分析和系统发育研究[J].中草药,2022,53(4):1127-1137
[27] Zhang T, Huang S, Song S,. Identification of evolutionary relationships and DNA markers in the medicinally important genusbased on chloroplast genomics [J]. PeerJ, 2021,9:e12612
[28] Chen Q, Hu H, Zhang D. DNA Barcoding and phylogenomic analysis of the genusin China based on complete chloroplast genomes [J]. Front Plant Sci, 2022,13:764255
[29] Dong W, Xu C, Li C,. ycf1, the most promising plastid DNA barcode of land plants [J]. Sci Rep, 2015,5:8348
Relationship between Chloroplast Genomes Structures fromPlantsand Phylogeny
MA Xiao-jun, GUO Zong-lei, WANG Xin-hong, XUAN Jing, TIAN Jing, LIU Huan, SUI Yuan-yuan, SHU Jing*
,250000,
It is difficult to distinguish between plants of thebecause of their morphological similarity. Characteristics and phylogeny of chloroplast genomes, can provide a theoretical basis for study of the phylogeny, molecular identification, and resource exploitation of the. Based on the published chloroplast genome sequences of 29species, their chloroplast genomes were analyzed using bioinformatics methods. The results showed that, the chloroplast genome is between 151058 bp and 152434 bp. There are 128 to 133 genes in the, with a stable number of rRNA-coding genes, and differences in protein-encoding and tRNA-encoding genes. Significant differences exist at the IRb/SSC boundary. The highest content of tandem repeats and mononucleotide. No gene rearrangements were detected by covariance analysis, reflecting the high degree of conservation in the chloroplast genome of the. Phylogenetic analysis based on the chloroplast genome indicates that thecan be divided into four major phylogenetic clades. Nucleotide polymorphism analysis of the chloroplast genome offound that the IR was more conserved than the single-copy regions, and the non-coding region was mutated more frequently than the coding region.
; chloroplast; gene; phylogeny
Q75
A
1000-2324(2022)05-0811-08
2022-09-24
2022-10-11
国家重点研发计划(2019YFD1000102);山东省良种工程(2019LZGC009);山东农业工程学院校企科研创新团队(YBZR202204)
马晓君(1997-),女,研究生,助教,研究方向:基因工程. E-mail:mxjun7@163.com
通讯作者:Author for correspondence. E-mail:shujing79@163.com
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
肝博士(2022年3期)2022-06-30 02:48:28
艺术品鉴(2021年12期)2021-05-25 16:31:36
Journal of Sport and Health Science(2019年6期)2019-11-26 07:30:53
基层中医药(2018年9期)2018-11-09 01:20:32
中成药(2017年5期)2017-06-13 13:01:12
广西林业科学(2016年1期)2016-03-20 05:33:01
中国野生植物资源(2014年4期)2014-03-29 07:26:14
癌变·畸变·突变(2014年2期)2014-03-01 04:39:42
食品科学(2013年6期)2013-03-11 18:20:13
