
沸石孔道限域亚纳米金属团簇催化剂研究进展
2023-01-11 14:11刘月明何鸣元
华东师范大学学报(自然科学版) 2023年1期
马 跃,徐 浩,刘月明,张 坤,吴 鹏,何鸣元
(华东师范大学 化学与分子工程学院 上海市绿色化学与化工过程绿色化重点实验室,上海 200062)
0 引 言
高效催化转化CO2、CO、CH4、CH3OH和HCOOH等C1分子及低碳烷烃脱氢制备高附加值氢气(H2)、C2和C3烯烃、芳烃、汽油、柴油、二甲醚和乙醇等化学品的合成路线,已成为学术界和工业界的高度关注的热点和研究重点.与石油资源相比,C1资源和低碳烷烃储量丰富,其有效利用对能源、环境和经济的可持续发展具有重要意义[1].长期以来,沸石及沸石负载的金属纳米催化剂被广泛应用于此领域中的重要反应过程,设计合成新型沸石负载的纳米金属催化剂一直是研究的重中之重.众所周知,金属纳米颗粒的尺寸是决定负载型金属催化剂性能的重要因素[2-3].近年来,利用孤立原子和由少量原子组成的金属团簇的多相催化技术丰富了我们对纳米金属催化剂结构反应关系的认识[4-5].金属纳米颗粒的粒径减小会大大增加暴露的活性位点,特别是亚纳米金属团簇,甚至是单原子金属催化剂,都显示出了优异的催化活性和选择性[6-9].然而,由于粒径的减小赋予了其大幅增加的表面自由能,材料制备和催化过程中较差的稳定性和严重的团聚烧结仍然是小尺寸金属纳米颗粒催化剂发展的主要“瓶颈”[6,10].将亚纳米金属团簇锚定在载体上,其表面金属原子可以通过化学键与载体相连,不仅有效地提高了催化剂中金属物种的稳定性,而且载体也可以反过来改变亚纳米金属团簇的结构和电子特性,从而影响这些亚纳米金属物种的反应性[11].与碳[12]、金属有机骨架材料[13]和氧化物[14]等开放或近开放结构相比,具有均匀多孔结构的沸石可以将亚纳米金属团簇牢牢地限制或封装在其晶体内.这些多孔沸石载体可以作为强大的宿主,将金属纳米团簇封装在内部的孔道中,限制其尺寸进一步增长,带来更高的稳定性和抗烧结性[15-22].
与其他负载型金属催化剂相比,这种沸石孔道限域的亚纳米团簇型金属催化剂在获得高原子利用率的同时,即使在恶劣的制备和催化过程中,从载体到活性金属都表现出极佳的热/水热稳定性,这大大减少了金属组分的用量投入及在循环过程中的浪费,属于绿色化学的范畴[20-22].此外,又因为沸石材料良好的约束效应和固有的酸/碱性质,沸石孔道限域的亚纳米团簇型金属催化剂可以兼具高稳定性、择形选择性及协同催化的优势,引起了学术界和工业界的广泛关注和研究[19-44].作为一种高效纳米催化剂,利用受限的亚纳米金属团簇与具有活性位点的多孔沸石骨架之间的协同效应,不仅可以显著提高催化性能,还可实现底物分子在接力催化过程中的高效转化,被广泛应用于甲烷芳构化等C1分子转化过程及低碳烷烃脱氢/脱氢芳构化等工业领域[23-28].近年来,这些沸石孔道限域的亚纳米金属团簇型催化剂也被应用于许多新兴的催化领域,如涉及甲酸、氨硼烷或肼产氢的化学能源储存,以及生物质衍生燃料和化学品的生产等[2,20-22,29].
普通浸渍法和离子交换法是目前最常用的制备沸石负载金属催化剂的方法,但这两种方法通常会导致金属颗粒大、分散不均匀、催化活性和稳定性不理想[45-48].为了克服这些缺点,国内外众多学者已开发出多种新的合成方法,包括配体保护法[20,22,29-30]、配体保护的直接H2还原法[31-32]、前驱体稳固法[17,33]、含金属晶种定向合成法[34]、浸渍-溶解-再晶化法[35]、沸石结构转变法[19,36],以及本课题采用的沸石骨架杂原子诱导锚定法[37-38]等,以上方法均能够将一种或几种金属以亚纳米团簇的形式很好地封装在沸石孔道内.近年来,随着新型电子显微镜(如球差校正高角度环形暗场扫描透射电子显微镜(Cscorrected high-angle annular dark-field scanning transmission electron microscopy,Cs-correct HAADF-STEM))、X射线吸收光谱(X-ray absorption spectroscopy)和探针分子红外光谱等表征技术的发展,研究者们对沸石孔道内限域的亚纳米金属结构和落位的认识变得更加全面且准确,这也极大地促进了沸石孔道限域金属催化剂的发展[46,49].
本文综述了最近几年较为典型的制备沸石孔道限域的亚纳米团簇型金属催化剂的方法及此类催化剂在几种重要的热点反应中的催化应用研究,包括CO2和炔烃选择性加氢[29,39-40]、甲酸和氨硼烷水解产氢[20-22,29-30],以及丙烷脱氢[19,32,37-38,41-44]等反应过程.
1 沸石孔道限域金属催化剂的制备方法
1.1 湿法浸渍
湿法浸渍是将催化剂的金属组分以固态方式装入多孔载体最简单的方法,目前可用于将亚纳米级金属团簇封装到沸石孔道中[48].一般的制备步骤是:首先将脱水、脱气的沸石与一定量的含金属前驱体的溶液混合在水或乙醇中,进行包覆浸渍.然后将得到的固体过滤、干燥,根据所用金属的不同在流动氢气或其他处理条件下进行焙烧[49].这种浸渍过程通常伴随着所用溶剂的不断蒸发,以促进金属前驱体在毛细凝聚力作用下扩散到载体孔隙中[21].但往往由于沸石中可能残留气体及溶液浸润性较差,金属前驱体难以扩散到沸石的内部孔隙中,导致金属团簇分布不均匀,尺寸较大的金属颗粒可能位于沸石的外表面而不是内部.值得注意的是,金属阳离子负载量对分散的均匀性也有显著影响.例如,Huang等[50]发现当Pd负载量为0.01 wt%(质量百分比,下同)时,Pd原子能够均匀地分散在沸石微孔中,而当负载量提高为2.0 wt%时,在ZSM-5晶体的外表面出现明显的Pd O颗粒.虽然浸渍法在过去并不是封装亚纳米金属团簇的理想方法,但是最近Wang等[21]通过简单的浸渍法在自柱撑MFI沸石(S-1和ZSM-5)纳米薄片中封装了亚纳米单金属Rh、Ru及各种RhxRu1-x双金属团簇(图1(a)).他们发现,Rh0.8Ru0.2双金属团簇的平均尺寸小于1 nm且被均匀分散在MFI的微孔内(图1(b)—(i)).而常规MFI沸石浸渍后的金属纳米颗粒尺寸较大,分布不均匀,主要分布在沸石外表面.单金属Rh、Ru及各种RhxRu1-x双金属团簇的成功封装主要是借助自柱撑MFI沸石的独特结构.与常规MFI沸石骨架相比,自柱撑MFI沸石具有显著增加的外表面,特别是具有丰富的缺陷位硅羟基,这有利于改善金属前驱体在沸石中的分散和固定.本课题组为了克服浸渍法会导致金属物种不均匀分散的缺点,也做了一些原创性的工作,利用沸石骨架杂原子Sn诱导锚定客体Pt物种的方法,制备了*BEA沸石孔道内限域的Pt亚纳米团簇[38].我们选择高骨架锡含量的Sn-Beta沸石(nSi/nSn=80,摩尔比,下同)为载体,为客体Pt提供足够的锚定位点;在含水条件下,载体Sn-Beta由封闭状态的四配位(Si-O)4Sn(Ⅳ)转变为六配位的开放状态(Si-O)3(H2O)2Sn(Ⅳ)-OH;其羟基与水溶液中的H2PtCl6反应形成催化剂前体Pt-Sn-Beta-P,Pt物种以单分散的(Si-O)3(H2O)2Sn(Ⅳ)-(O)-[PtCl5]-形式存在;最后经过H2直接还原,制备得到Sn-Beta沸石孔道封装Pt亚纳米团簇的催化剂Pt@Sn-Beta (图1(j)).Cscorrect HAADF-STEM表明,尺寸分布在0.2~1.0 nm的亚纳米Pt团簇被牢牢地封装在Beta沸石孔壁中,并且与骨架Sn之间以Pt-O-Sn键形式连接.Wang等[37]也报道了类似结果.此外,Wei等[51]还发现,通过使用柠檬酸络合策略,将柠檬酸镍水合物与硝酸铈同时浸渍到沸石中,可以实现Ce和Ni的亚纳米级高度分散.
1.2 离子交换
近年来,离子交换法被广泛应用于将金属物种作为催化活性位点引入沸石分子筛孔道中.它通常将含有金属前驱体稀溶液中的一个离子与沸石中的另一个离子交换.大多数情况下,该工艺只需几个小时就可在室温下完成金属前驱体装入沸石,但这往往导致载量较低.为了提高离子交换量,通常需要多次重复离子交换步骤,并适当提高温度以提高交换程度[46-47].即使在离子交换步骤后,过滤和洗涤多余的离子时也必须严格控制该操作溶液的pH值,以防金属阳离子在沸石的外表面预沉淀.为了调节金属团簇在沸石中的分散,后续焙烧后的处理步骤至关重要,该步骤通常包括金属前驱体的氧化和还原.Graaf等[52]指出,焙烧步骤的升温速率对封装有显著影响.在焙烧过程中,Pt(NH3)42+中的氨配体以较低的速率被氧化为N2O和H2O.如果加热速度过快(>0.2℃/min),氨配体会部分氧化,从而导致尺寸较大的Pt纳米颗粒形成.Moliner等[53]研究发现,Pt前驱体可以通过离子交换的方式扩散到硅铝摩尔比约为8的硅铝型CHA沸石中,并实现亚纳米Pt物种的封装而不团聚(图1(k)),这与水热法制备的材料[54]几乎没有区别.有趣的是,他们还发现Pt团簇和单原子分别在H2和O2中能够可逆地相互转换.然而,这并不是一个普适性的结果,当以类似硅铝摩尔比的ZSM-5为载体时,负载的Pt不具备足够的稳定性.目前,CHA结构中导致这种优异的原子俘获特性的结合位点尚未可知.
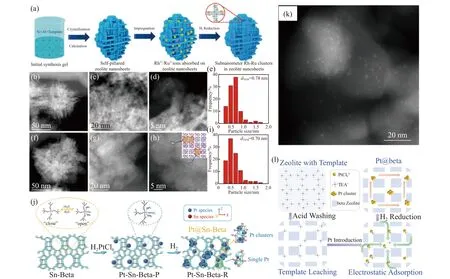
图1 浸渍、离子交换和模板剂引导法制备沸石孔道限域型金属催化剂Fig.1 Zeolite-confined metal catalysts prepared by impregnation,ion exchange,and template-guided methods
1.3 模板剂引导
最近,Tian等[55]开发了一种模板剂引导的方法,以避免在外表面形成大块金属颗粒.与浸渍法和离子交换法需要焙烧去除模板完全不同,此方法是使用有机酸(如醋酸)将合成样品的结构导向剂部分去除,从而形成金属离子的扩散通道.剩余带正电荷的模板剂(如TEA+)可以通过静电作用锚定带负电荷的PtCl6—,而不需要使用有机配体.模板剂在后续的H2直接还原过程中逐渐分解,并伴随着在沸石孔道的连接处封装亚纳米金属团簇(图1(l)).总体而言,研究者通过合理设计沸石的形貌和骨架、金属配体的功能及沸石模板与金属前驱体之间的静电相互作用等,创制了全新的后修饰策略以将亚纳米金属团簇封装在沸石中.此方法制备的沸石限域金属催化剂Pt@H-beta,由于其Pt物种高度分散,与沸石相互作用强,促进了氢从Pt到沸石的转移,使得其具有较高的酚醛加氢脱氧性能及优越的催化稳定性.
1.4 原位水热合成
原位水热合成是合成沸石最广泛采用的方法[56],通常包括成凝胶过程和随后的水热结晶过程.为了封装金属位点,将含有硅源(和铝源)及直接合成沸石所需模板剂的原始凝胶与金属前驱体(通常为氯化物和硝酸溶液)混合在一起[16,22,30,57].然后在自身压力下进行数天的水热结晶过程,再经过后续的空气焙烧和H2还原,最终得到沸石封装金属催化剂.在水热合成沸石过程中,较高的合成温度和p H值通常会促进金属前驱体的析出,形成胶态氢氧化物,不利于金属前驱体向沸石内部孔隙扩散,所以一般需要配体的辅助[22].Sun等[20]以[Pd(NH2CH2CH2NH2)2]Cl、Co(NH2CH2CH2NH2)3](NO3)2和Ni(NH2CH2CH2NH2)3(NO3)2等金属乙二胺配合物为前驱体,在170℃水热条件下晶化3 d,制备了MFI结构全硅分子筛S-1封装的亚纳米双金属团簇Pd-M(OH)2(M为Ni,Co)(图2(a)).Cs-correct HAADF-STEM和EXAFS(extended X-ray absorption fine structure)结果显示,亚纳米双金属Pd-Ni(OH)2团簇的平均直径大约为1 nm,Pd-Pd配位数仅为2.3~3.1(图2(b)—(c)).封装于S-1沸石中的金属团簇的尺寸远小于通过浸渍法形成的双金属纳米颗粒.此过程不仅没有破坏沸石骨架,受限的双金属团簇还被固定在了MFI结构的直型通道和正弦通道之间的交叉处.在此水热合成的基础上,Sun等[31]又开发了一种不经空气焙烧,配体保护下直接H2还原的方法,将单个Rh原子成功封装在MFI沸石的正弦孔道五元环内,并由沸石骨架氧原子来稳定(图2(d)—(e)).在H2直接还原过程中,有机配体和模板作为金属保护剂,随着金属Rh物种的还原而逐渐分解.他们还将此方法应用于封装双金属亚纳米Pt-Zn团簇到S-1沸石中(图2(f)),有效地防止了常规煅烧-还原过程中亚纳米金属团簇的烧结[32].Cs-correct HAADF-STEM结果表明,该方法制备的PtZn@S-1-H中的PtZn双金属团簇均匀分布在S-1沸石的直型和正弦通道(图2(g)—(h)).如图2(i)—(k)所示,Liu等[42]在MFI合成体系中同时添加Pt源和Sn源,一锅法合成了PtSn-MFI材料.又通过在合成凝胶中引入K+,最终得到的K-PtSn@MFI很好地抑制了空气煅烧过程金属的聚集,获得的亚纳米Pt团簇选择性落位在MFI结构的正弦通道.

图2 原位水热法制备沸石孔道限域型金属催化剂Fig.2 Zeolite confined metal catalysts prepared via the in-situ hydrothermal method
1.5 沸石结构转变
早在多年以前,Goel等[36]就通过沸石间转化的方法将载有金属的*BEA或FAU沸石成功转变为限域金属团簇(Pt,Ru,Rh)的MFI分子筛(图3(a)).与直接水热合成方法相比,沸石间转化大大减少了合成沸石的时间和成本,并可以通过添加晶种来避免有机结构导向剂的使用,这是一种更经济和环保的方法.除了这种在三维沸石之间发生结构转换的方法,利用片状二维沸石和三维沸石之间的转换是另一种极具潜力的亚纳米金属团簇的封装策略.最近Liu等[19]报道了通过2D沸石转化为3D沸石的过程,将少量Pt原子和亚纳米团簇封装到MCM-22沸石的超笼中.如图3(b)所示,先用有机表面活性剂十六烷基三甲基铵(CTMA+OH—)结构“柔性”的全硅片状MWW前驱体进行溶胀,亚纳米Pt物种会纳入单个MWW层之间的内部通道中,最后经过空气焙烧将Pt原子和团簇封装在MCM-22沸石的“杯”和笼中.Pt负载量为0.11 wt%,且Pt原子和团簇高度分散(图3(c)).在650℃下经过4次氧化还原处理后,团簇尺寸仍能保持在2 nm以下.而湿浸渍法制备的Pt/MCM-22-imp催化剂的Pt尺寸在相同的条件下显著增加到30~50 nm.采用相同的方法,他们又将Au亚纳米团簇封装到MCM-22内部(Au@MCM-22-S),但Au含量较低仅为0.025 wt%[58].为了将Au的担载量提高,他们将1-辛硫醇加入溶胀液中(图3(d)).由于Au-S键的强相互作用,所得Au@MCM-22-L催化剂的Au负载量提高到0.11 wt%.然而,伴随着Au@MCM-22-L的高Au负载量,导致了Au亚纳米团簇和纳米团簇(约1 nm)的同时出现(图3(e)—(h)).

图3 沸石结构转变法制备沸石孔道限域型金属催化剂Fig.3 Zeolite confined metal catalysts prepared by the interzeolite transformation method
2 沸石孔道限域金属催化剂的催化应用
2.1 选择性加氢反应
沸石包裹的亚纳米金属团簇在加氢反应中表现出良好的催化性能.Wang等[39]报道了一种利用含金属晶种定向合成法,此法制备了沸石晶体包裹的金属Rh催化剂,通过改变沸石种类(ZSM-5、S-1、S-1-OH和KZSM-5)来控制CO2加氢过程中产物的选择性.如图4(a)—(f)所示,几种不同特性的MFI沸石封装的Rh催化剂均具有较高的CO2转化活性. 其中,Rh@S-1倾向于生成CO,而Rh@ZSM-5却表现出极高的CH4选择性,这为CO2选择性加氢催化剂的设计提供了新的思路.Sun等[29]在原位水热条件下,采用配体保护的方法将亚纳米双金属Pd-Mn团簇封装在S-1分子筛中.与浸渍法制备的Pd/S-1-im及单一金属的Pd@S-1相比,由于形成了细小且高度分散的金属团簇,以及双金属组分之间的协同作用,Pd Mn@S-1催化剂上CO2加氢生成甲酸酯的速率有所提高(图4(g)).其中,Pd Mn0.6@S-1具有最高的CO2加氢活性和良好的稳定性(图4(g)—(h)).沸石包裹的亚纳米金属团簇除具有极佳的CO2加氢催化效果外,在炔烃不完全加氢反应上也有较为理想的表现.例如,Wang等[40]发现封装在SOD分子筛中的亚纳米Pd团簇(Pd@SOD)在乙炔半加氢制乙烯过程中表现出优异的催化选择性(高达94%),而浸渍的Pd/SOD只有21.5%左右(图4 (i)).他们认为,这种独特的封装结构是高催化选择性的关键.一方面,SOD沸石的孔隙结构只能允许氢分子进入,然后被包裹的Pd团簇活化,通过氢溢流过程形成活性OH物种,反应物乙炔与SOD外表面的OH物种反应得到所需的乙烯.另一方面,SOD分子筛的孔结构严格阻碍了乙烯与Pd团簇的直接作用,避免了过度氢化.这一结论也被DFT计算所证实(图4(j)).此外,Liu等[19]利用沸石结构转变法制备的MCM-22封装的亚纳米Pt团簇(Pt@MCM-22)与浸渍Pt/MCM-22催化剂相比,在丙烯加氢反应中表现出更高的催化活性.

图4 不同分子筛限域金属催化剂的选择性加氢性能Fig.4 Performance of various zeolite confined metal catalysts in selective hydrogenation reactions
2.2 甲酸或氨硼烷产氢
氢气是目前最有前景的清洁能源之一[59-60].然而,由于氢的物理特性,其运输、储存和释放仍然面临着巨大的挑战.寻找安全高效的储氢材料以转型到氢能社会是当前氢能应用面临的最大挑战之一[61].因为甲酸(formic acid,FA)和氨硼烷(ammonia borane,AB)在储氢和放氢性能方面具备的显著优势,使得FA分解制氢(HCOOH→H2+CO2)和AB水解制氢(NH3BH3+2H2O→NH4++BO2—+3H2)成为当前的研究热点.Sun等[20,29]和Wang等[21-22]在这一领域已做了大量细致的研究.例如,他们以[Pd(NH2CH2CH2NH2)2]Cl2为前驱体,在直接水热条件下成功制备了包裹在纳米S-1沸石孔道交叉处的超细Pd团簇.在温和的反应条件下,制备的催化剂具有良好的活性,能够高效产氢而不生成CO,使FA在体系中完全分解.此外,由于Pd团簇在沸石基体内的有效约束,合成的催化剂在FA分解过程中具有良好的热稳定性和循环稳定性[22].之后,他们又在合成体系中添加了另外一种非贵金属(Co,Ni),制备得到了亚纳米级非贵金属氢氧化物杂化的双金属团簇,并被牢牢地封装在纳米级纯硅沸石S-1内部[20].其中0.8Pd0.2Ni(OH)2@S-1催化剂表现出良好的催化活性和高效的产氢能力.需要特别指出的是,其初始TOF(turover frequency)值高达5 803 h—1,约为传统Pd/C催化剂的16倍(图5(a)—(b)).此TOF值几乎是相似条件下非均相催化FA分解活性最高的催化剂,甚至高于大多数均相催化剂[62-63].与0.8Pd0.2Ni(OH)2@S-1催化剂相似,他们制备的Pd Mn0.6@S-1催化剂在FA分解中也表现出了优异的催化活性[29],即60℃温度下的初始TOF值为6 860 molH2·molpd—1·h—1,而且不生成CO.更重要的是,由于Pd-Mn亚纳米簇被封装及此双金属组分之间的协同作用,相比单金属Pd@S-1而言,Pd Mn0.6@S-1催化剂大大提高了产氢速率且连续运行5次后仍保持不变,表现出极高的循环稳定性(图5(c)—(d)).除FA的分解可以产氢外,近年来AB的水解产氢也备受关注.Wang等[21]通过简单的湿法浸渍在自柱撑ZSM-5沸石纳米薄片中封装了亚纳米RhxRu1-x双金属团簇,所得催化剂具有良好的AB水解性能.这归因于ZSM-5沸石的双金属团簇与Brönsted酸位点之间具有高效的协同作用,可显著促进水分子的活化,加速AB水解产生H2.其中Rh0.8Ru0.2/SP-ZSM-5-100催化剂表现出极高的催化活性,在3 min内使AB完全分解,产生73.5 mL的H2(nH2/nAB=3),在25℃和40℃下TOF值 分 别 为1 006 molH2·molmetal—1·min—1和3 001 molH2·molmetal—1·min—1(图5(e)).该TOF值 比Rh/Nano S-1的高15倍,比Rh0.8Ru0.2/SP-S-1的高2.5倍;与报道的其他多相催化剂相比,几乎是最高的.此外,该自柱支撑ZSM-5沸石封装的双金属催化剂表现出良好的循环使用寿命(图5(f)),连续5次循环后,H2生成速率仍然能够达到812 molH2·molmetal—1·min—1的高TOF值,同时金属团簇大小和沸石形态均保持不变.
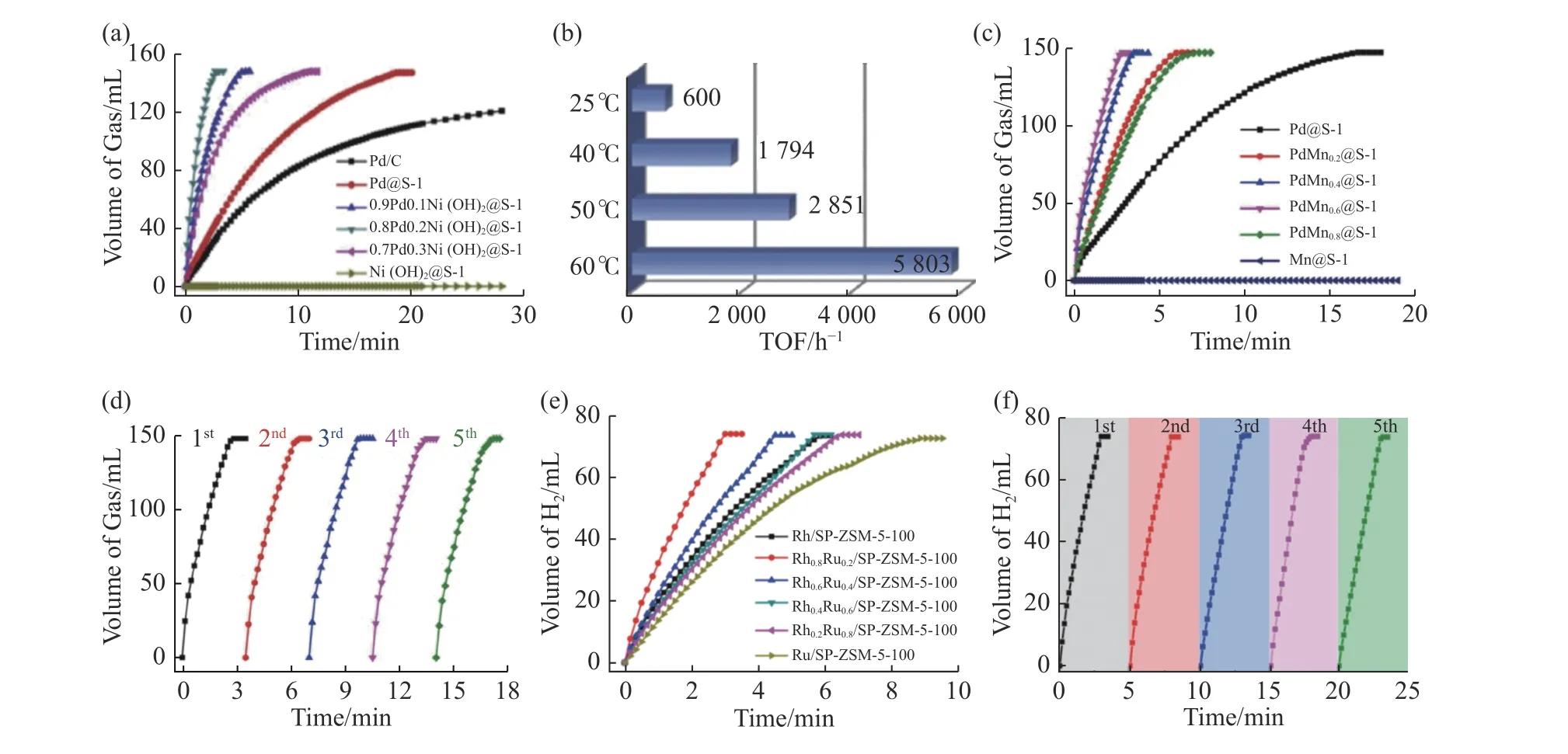
图5 不同分子筛限域金属催化剂的甲酸或氨硼烷产氢性能Fig.5 Performance of various zeolite confined metal catalysts in hydrogen generation by formic acid decomposition and ammonia borane hydrolysis
2.3 丙烷脱氢
近年来,由于丙烯需求的逐年增长和页岩气产量的增加,丙烷脱氢引起了广泛关注.Pt基负载型催化剂倾向于活化丙烷中的碳氢键,而非碳碳键.因此,Pt基负载型催化剂具有较佳的丙烷脱氢效果,Pt也是目前唯一一个商业应用的贵金属[64].然而Pt基负载型催化剂仍有其使用局限:一方面,高价格的贵金属Pt无疑会增加投入成本;另一方面,由于丙烷脱氢是强吸热反应,化学平衡常数小,需要较高的反应温度,这就使得反应过程中Pt颗粒容易发生团聚、烧结,导致催化剂迅速失活,产物选择性下降.因此,采用具有均匀微孔结构的沸石做载体,以及添加第二种金属助剂制备限域型单/双金属亚纳米团簇催化剂目前成为此领域的研究热点[19,32,37-38,41-44].例如,Liu等[42]过引入K+稳定Pt物种和Sn物种,一锅法制备了MFI孔道限域的PtSn双金属亚纳米团簇催化剂(K-PtSn@MFI),在催化丙烷脱氢反应中获得了高的丙烯选择性(约90%)和丙烷转化率(大于60%)(图6(a)).更重要的是,K-PtSn@MFI这种高催化活性至少可以维持3个反应-再生循环而不发生明显的失活,且亚纳米Pt Sn物种的大小和空间位置保持不变.Sun等[32]采用一种配体保护的直接氢还原方法,制备的MFI沸石封装的双金属PtZn合金型亚纳米团簇催化剂大大改善了丙烯选择性和催化稳定性.其中,PtZn4@S-1-H催化剂在质量小时空速(weight hourly space velocity,WHSV)为3.6 h—1时,丙烷转化率接近50%,丙烯选择性高达99.3%(图6(b)—(c)).即使在运行13 000 min后,PtZn4@S-1-H催化剂上也没有观察到明显的失活(图6(d)),失活常数极低,为0.001 h—1.此外,他们发现,Cs+离子引入沸石通道可以有效抑制金属向外表面迁移和聚集,可以进一步提高催化剂的稳定性.PtZn4@S-1-H-Cs催化剂的丙烷转化率和丙烯选择性在整个反应过程中,甚至在连续4次再生循环后都没有明显变化(图6(e)).几乎同一时间,Wang等[43]采用简单的原位合成方法也在S-1沸石中封装了分布均匀的超细PtZn双金属纳米团簇,其中0.3Pt0.5Zn@S-1催化剂在丙烷脱氢反应中具有出色的催化性能.与众多原位合成方法不同,本课题组另辟蹊径[38],利用沸石骨架Sn诱导锚定客体Pt物种,采用简单的浸渍即制备得到Beta沸石孔道封装的Pt亚纳米团簇,在丙烷脱氢反应中展现高活性,高丙烯选择性以及高稳定性.如图6(f)—(g)所示,丙烷的初始转化率(47%)维持了超过85 h,丙烯选择性高达99%.410 h后,丙烷转化率仅降低10%,丙烯选择性基本不变,在经历提高反应温度(图6(h)点A)以及对失活催化剂进行氧化再生(图6(h)点B)这样一个整体的过程中,Pt-Sn-Beta-R催化剂能够稳定催化丙烷脱氢反应近800 h,表明该催化剂具有突出的丙烷脱氢性能.Wang等[37]也报道了类似结果.
3 总结和展望
随着金属纳米粒子尺度的减小,其能级结构会发生急剧变化,使得金属纳米团簇甚至单原子产生独特的性能,近年来在催化领域得到广泛研究和应用.但是,随着尺寸减小而增大的表面自由能使亚纳米金属团簇在稳定性上也有着明显的缺陷,主要表现为易团聚、高温烧结、热稳定性差、金属流失、循环使用次数少等.故可以采用热/水热稳定且具有均匀微孔结构的沸石对亚纳米金属团簇进行封装.此类催化剂具有比传统催化剂更高的原子利用率和稳定性,减少了催化剂的一次性投入和循环浪费,符合绿色化学的宗旨.不仅如此,沸石骨架上的某些特殊活性位点,如Al、Sn,甚至Si-OH缺陷位等能够与限域的亚纳米团簇进行协同,从而提升催化活性或者产生新的催化性能.以沸石材料为载体的孔道限域型亚纳米团簇催化剂在多相催化领域正发挥着越来越重要的作用,在C1分子转化、低碳烷烃脱氢及氢能源的储存与释放等领域具有重要应用潜力.为了满足实际应用多样化的需求,需要开发新颖和简单的封装策略制备性能优异且高度稳定的沸石孔道限域的亚纳米金属团簇型催化剂.此外,未来应考虑跨学科整合,基于沸石孔道限域的亚纳米团簇型金属催化剂开发更重要、更有价值的催化反应过程和工艺.沸石孔道限域的亚纳米团簇作为一类重要的多相催化剂,有望在实际工业生产中得到应用和发展.
猜你喜欢
潍坊学院学报(2021年2期)2021-07-22
航天工业管理(2020年9期)2020-12-28
重型机械(2020年2期)2020-07-24
山西化工(2019年4期)2019-02-17
石油石化绿色低碳(2019年6期)2019-01-14
石油化工建设(2018年2期)2018-07-11
山东化工(2018年10期)2018-06-07
物理化学学报(2018年1期)2018-01-29
浙江工商大学学报(2015年3期)2016-01-07
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
