
制革废水中含铬有机物的生物吸附-矿化协同去除机制
2023-01-04 01:21:14党靖雯马宏瑞朱超徐芝芬周姣
皮革科学与工程 2023年1期
党靖雯 ,马宏瑞 *,朱超 ,徐芝芬 ,周姣 ,2
(1. 陕西科技大学环境科学与工程学院,陕西 西安 710021;2. 桂林理工大学环境科学与工程学院,广西 桂林 541004)
引言
制革含铬有机废水主要来源于主鞣、复鞣、染色和加脂阶段,其过程中会加入以甲酸为代表的低分子有机酸类、以多酚为主的单宁类植物鞣剂、以羧基为主的丙烯酸类鞣剂等[1],这类物质会与铬配位形成线性、网状等稳定性极强的有机物-铬络合体(OM-Cr)[2-3]。
制革废水经常规“物化+生化”工艺处理后,生化尾水仍含有0.3~0.5 mg/L[4]的总铬。研究表明,加碱沉淀后稳定于水相的铬主要以络合态形式存在[5],残余Cr(III)通过羟基- 羧基键合形成低分子态配合物[6]。这部分有机络合态铬,会进入综合废水处理阶段[7],在生化处理时吸附在活性污泥中[8]。
活性污泥的胞外聚合物(EPS)主要组分为蛋白质、多糖、脂类等[9-10],因含有亲水和疏水基团使其可通过静电相互作用和离子交换作用吸附重金属离子[11],同时EPS 上的羧基、羟基等基团也可与金属离子形成配合物[12-13]。制革废水生化处理过程中EPS 与Cr(III)的相互作用并非是单一对离子态铬的吸附[14],可能存在低分子酸络合态Cr(III)与EPS 的竞争络合作用[15]。目前有关制革废水中络合态Cr(III)与活性污泥EPS 的相互作用及其稳定性的研究报道较少,由此导致铬达标不稳定的原因尚不清晰。
本文对制革废水生化处理中活性污泥吸附废水中的铬进行了模拟,以此为案例,通过探究活性污泥对含铬有机废水的吸附过程,对比分析吸附前后有机物与Cr 的协同分布特征,分析活性污泥EPS 吸附前后Cr(III)在各结构层的分布量及各组分变化,结合吸附前后的傅里叶红外光谱仪(FT-IR)、三维荧光光谱(3D-EEM)测试,进一步分析生化破络过程,识别污泥相中的Cr(III)稳定化程度,以期为制革废水铬排放总量控制及深度处理提供理论依据。
1 材料与方法
1.1 材料
制革复鞣染色段废水取自江苏省某制革企业现场的复鞣染色段废水,其主要污染物指标见表1,活性污泥取自西安市第五污水处理厂二沉池,曝气10 min 作为初始吸附样品。
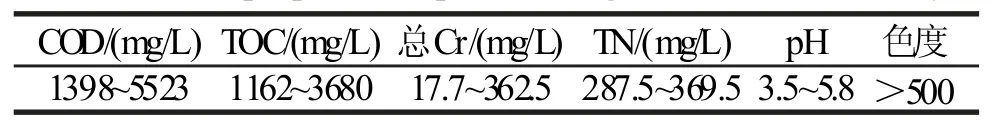
表1 某制革企业染整废水的基本性质Tab. 1 Basic properties of post-tanning wastewater of tannery
所用主要试剂考马斯亮蓝试剂、蒽酮等购于上海瑞楚生物科技有限公司,阳离子交换树脂购于上海开平树脂有限公司,微孔滤膜为醋酸纤维素滤膜、超滤膜为聚醚砜(PES)均购买自上海摩速科学器材有限公司。
1.2 实验方法
1.2.1 生化处理前后废水中有机物与Cr 的去除及粒径分布特征
取企业污水站好氧生化池活性污泥静止沉淀,置于1.5 m3模拟生化反应器(长×宽×高=1.8 m×1 m×1 m)中,确保污泥沉降体积(Sludge Settling Velocity 30 min,SV30)=50%。取车间含铬染色废水调节其pH 至8.5~9.5,调节进水流速进入反应器,水力停留时间(HRT)= 24 h 监测出水化学需氧量(COD)和总铬。分别进行6 个批次的实验,实验周期总计144 h,24 h 处理后水样进行滤膜过滤,微滤膜孔径分别为0.45、0.22 和0.1 μm,超滤膜采用截留相对分子质量为1~50 kDa 的滤膜,详细方法参见文献[16],测定不同滤液中总铬、总有机碳(TOC)和总氮(TN)含量。对第6 批次实验每隔2 h 取样,直到吸附时间达16 h,监测COD 和Cr(III)随时间的变化趋势。
1.2.2 活性污泥对废水中铬的吸附及其在污泥EPS结构层的分布
取适量活性污泥与染色水混合为污泥浓度沉降比(SV30)=50%,曝气吸附2 h,待活性污泥沉降至泥水比为1∶1 时,测其上清液中COD 和Cr,置去上清液,控制泥水比为1∶1 继续加入染色水,吸附第2 次,待活性污泥沉降至泥水比为1∶1 时,测其上清液中COD 和Cr,以此类推,共进行5 次吸附实验,测其上清液COD 和总Cr。
取生化曝气144 h 后的活性污泥进行分离提取可溶性 EPS (S-EPS)、松散型结合态 EPS(LB-EPS)和紧密型结合态 EPS(TB-EPS),测定并分析各结构层EPS 提取液中的Cr 浓度及EPS 组分的含量变化。采取改良热提取法[17]对EPS 进行提取预处理,控制温度≤80 ℃,加热时间≤60 min;采用“差速离心法+阳离子交换树脂法”提取好 氧 活 性 污 泥 S-EPS、 总 EPS、TB-EPS 与LB-EPS,该方法对铬在EPS 各结构层的分布造成的影响较小[18-19]。
1.3 分析方法
总铬采用高锰酸钾氧化-二苯碳酰二肼分光光度法(GB/T 7466-1987)测定;多糖(PS)采用蒽酮- 硫酸法测定;蛋白质(PN)采用考马斯亮蓝法测定;TN 采用碱性过硫酸钾紫外分光光度法(HJ 636-2012);pH 采用 pH 计测定;COD 采用 COD 快速测定仪(5B-3F 北京连华)测定。
采用 TOC 分析仪(Liqui TOC II,德国 Elementar)测定溶解性有机碳(DOC)浓度,傅里叶变换红外光谱仪(FI-TR,Vertex 70 德国 Bruker)分析反应前后官能团变化,3D-EEM(爱丁堡FS5,英国Edinburghinstrume)测定采用150 W 氙灯作为稳态光源;PMT 电压为 400 V;发射波长(Em):225~550 nm;激发波长(Ex):220~500 nm;狭缝宽度 3.2 nm;带通Em 为 1 nm,Ex 为 5 nm;响应时间 0.1 s,将水样统一稀释10 倍,以超纯水为空白样。
2 结果与讨论
2.1 好氧生化处理对含铬有机废水COD 和Cr(III)的去除过程
图1 为好氧曝气24 h对不同批次废水中COD和Cr(III)的去除情况。由图可知,进水水质波动较大,HRT 为 24 h 时,COD 的质量浓度达 2407~777 mg/L,Cr(III)质量浓度达 30.0~5.6 mg/L。COD 和 Cr(III)的去除率分别达38.6%~56.85%和60.3%~69.2%,即使如此,生化尾水中Cr(III)质量浓度仍然有5~10 mg/L。

图1 不同批次废水好氧生化24 h 处理前后COD 和Cr(III)的削减效果Fig.1 Reduction effect of COD and Cr(III) before and after 24 h aerobic biochemical treatment in different batches of wastewater
图2 分别表示含铬有机废水原水和活性污泥吸附24 h 后废水上清液中TOC、TN 和 Cr(III)尺寸分布。本研究主要关注溶解性Cr(III),后续主要分析小于450 nm 的部分。由图2(a)可知,原水中TOC 集中在大于450 nm 颗粒态有机物和小于8 nm 的溶解态有机物,分别占总TOC 的26.56%和48.67%;Cr(III)集中在 2~3 nm 和小于 2 nm 的溶解态,分别占Cr(III)的 14.3%和 72.4%;TN 分布与TOC 类似,分别占总TN 的30.98%和51.59%,说明原水中含有大量有机络合态铬。由图2(b)可知,经24 h 生化处理后的水样,小于8 nm 的溶解态有机物TOC 削减率达到 77.71%,C/N 比由原水的 7.00~20.9 降至4.2~5.7;Cr(III)的分布与原水样相似,集中在小于5 nm 的部分,削减率达到68.55%,与图2(a)相比,大于450 nm 部分无不溶性含Cr(III)物质,各级区域内Cr(III)含量均降低且低于5 mg/L,说明活性污泥将不溶性含Cr (III) 物质完全吸附;Cr (III)、TN 与TOC 的分布类似,说明络合态铬部分被吸附,经过吸附矿化后上清液中残留的少量Cr(III)为有机络合态铬。
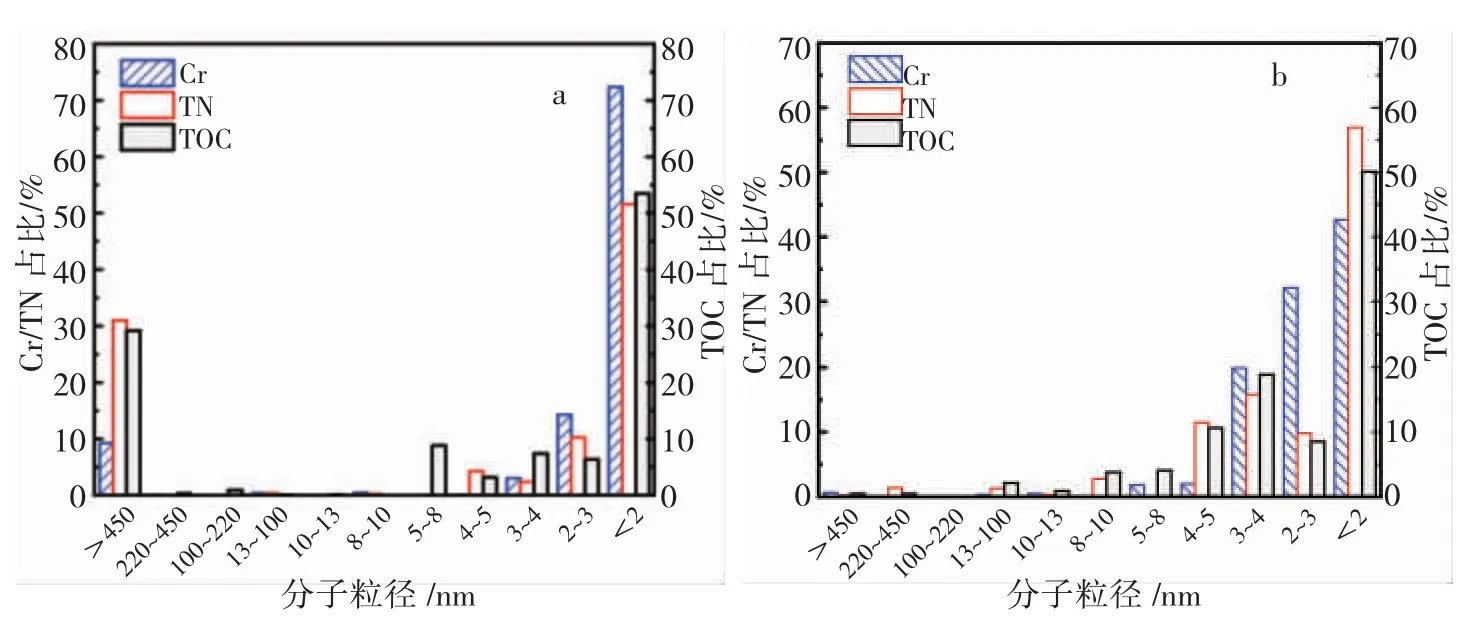
图2 生化处理前后水样中Cr(III)、TN 和TOC 的尺寸分布占比(a:原水;b:24 h 生化后)Fig.2 Size distribution ratios of Cr(III), TN and TOC in wastewater samples before and after biochemical treatment (a: raw water; b: 24 h treatment sample)
图3 为好氧活性污泥对废水中COD 和铬的吸附测定结果,COD 和Cr (III) 的去除率分别达46.7%~52.2%和 77.3%~83.4%,废水中的 Cr(III)的质量浓度从57.7 mg/L 降至10 mg/L 以下。持续16 h后,COD 浓度无明显变化,活性污泥对Cr(III)持续性吸附且Cr(III)质量浓度降至3 mg/L 后保持稳定,去除率达94.8%,多次重复实验效果基本一致。
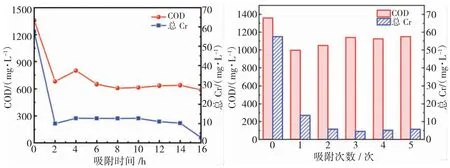
图3 好氧活性污泥对废水中COD 和铬的吸附(a:单次;b:多次)Fig.3 Adsorption of COD and chromium in wastewater by aerobic activated sludge (a: single; b: multiple)
为进一步确认活性污泥对铬的持续性吸附能力,实验采用吸附后的活性污泥对原水进行多次吸附,结果如图3 所示。由图3(b)可知,活性污泥1 次吸附后COD 降到910 mg/L,从第2 次吸附起,COD浓度保持在1100 mg/L 左右,削减率不足15%,随吸附次数增加,COD 浓度逐步接近原水浓度值,活性污泥对COD 的吸附基本达到饱和;从废水中Cr(III)的浓度削减变化来看,每一次吸附,Cr(III)均持续性降低,铬的质量浓度均从原水57.7 mg/L 降低到10 mg/L 以下,且每次吸附后铬的浓度波动变化不大,说明在一定范围内,活性污泥对Cr(III)可以持续吸附。好氧活性污泥在2 h 内可对COD 和Cr(III)发生同步吸附。
Ryszard 等[20]提出,活性污泥吸附重金属有机配体时生物细胞分泌的胞外聚合物与有机配体中的有机物竞争时占优势,推测随时间增长活性污泥降解COD,有机络合态铬破稳,释放出有机络合态中的铬离子被活性污泥吸附。
2.2 重金属铬对活性污泥EPS 组分的影响
2.2.1 活性污泥EPS 结构层中重金属铬分布量
图4 为活性污泥连续24 h 暴露在了不同浓度的 Cr(III)中,铬在各结构层 EPS,S-EPS、LB-EPS 和TB-EPS 中的分布情况。当Cr(III)质量浓度为5 mg/L时,TB-EPS、LB-EPS 和 S-EPS 提取液中铬质量浓度分别为 0.08、0.08 和 0.038 mg/L;当 Cr(III)质量浓度为 30 mg/L 时,TB-EPS、LB-EPS 和 S-EPS 提取液中铬质量浓度分别为0.174、0.196 和0.101 mg/L。由此可得,活性污泥暴露在不同浓度的铬下,各组分EPS中的铬分布呈相似规律,其较多分布在 TB-EPS 和LB-EPS 中且铬含量相当,S-EPS 中较少。各结构层EPS 理论和实际EPS 测量值的差值在±0.0038~0.015 mg/L 范围内,猜测有少部分铬进入细胞内,吸附在污泥EPS 上的络合态Cr(III)会重新分布、络合。
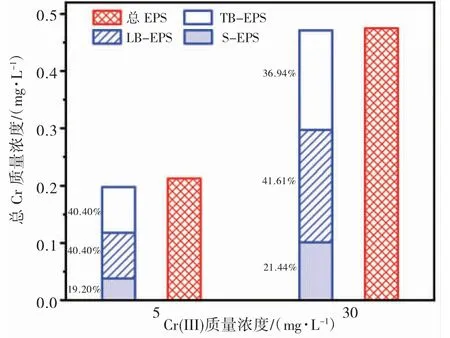
图4 Cr(III)在EPS 各结构层的分布情况Fig. 4 Distribution of Cr(III) in each structural layer of EPS
2.2.2 各结构层EPS 中多糖、蛋白质含量
图5 为活性污泥暴露在不同质量浓度Cr(III)的染色水中,各结构层EPS 中蛋白质和多糖的质量浓度5 mg/L 和30 mg/L 铬暴露下,EPS 质量浓度分别为 93.17 mg/L 和 73.91 mg/L,S-EPS 和 LB-EPS 中多糖为主要组分,蛋白质未检出。5 mg/L 铬暴露条件下,TB-EPS 中蛋白质为主要成分,质量浓度为19.18 mg/L。30 mg/L 铬暴露条件下,TB-EPS 中多糖质量浓度为9.93 mg/L,蛋白质含量为8.2 mg/L。
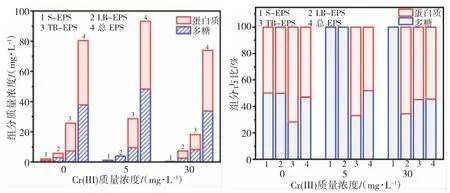
图5 不同质量浓度Cr(III)暴露下EPS 各结构层组分变化(a:组分质量浓度;b:组分占比)Fig. 5 Composition changes of EPS structural layers under exposure to different mass concentrations of Cr(III) (a: component mass concentration; b: component proportion)
综上可知,低浓度铬刺激污泥中微生物细胞分泌EPS,各结构层EPS 蛋白、多糖含量均升高,高浓度铬抑制微生物EPS 分泌,多糖含量减少量较显著,TB-EPS 中多糖含量随Cr(III)浓度的增大而升高。
2.3 吸附前后活性污泥EPS 光谱分析
2.3.1 三维荧光光谱分析
根据Wang[21]的研究,EPS 提取物中各种溶解性有机物的三维荧光光谱出峰位置可划分5 个区域,Ⅰ:Ex为 350~440 nm,Em为 430~510 nm;Ⅱ:Ex为 310~360 nm,Em为 370~450 nm;III:Ex为 260~290 nm,Em为 300~320 nm; Ⅳ :Ex为 230~290 nm,Em为320~350 nm;Ⅴ:Ex为 240~270 nm,Em为 370~440 nm。其中I 为类腐殖酸峰,III 和Ⅳ分别为类酪氨酸峰和类色氨酸峰,II、Ⅴ为类富里酸峰。
图6 和图7 表示活性污泥LB-EPS 和TB-EPS吸附铬前后的三维荧光光谱情况。由图可知,LB-EPS 含有四个荧光峰,峰A(Ex/Em=340/440)为类腐 殖 酸 峰 , 峰 B (Ex/Em=290/330) 和 峰 D(Ex/Em=230/340)为类色氨酸峰, 峰 C(Ex/Em=240/430)为类富里酸峰。TB-EPS含有五个荧光峰,峰A (Ex/Em=340/440)为类腐殖酸峰,峰B(Ex/Em=310/370)和峰C (Ex/Em=260/370)为类富里酸峰,峰D(Ex/Em=260/310)为类酪氨酸峰, 峰 E(Ex/Em=240/310)为类色氨酸峰。
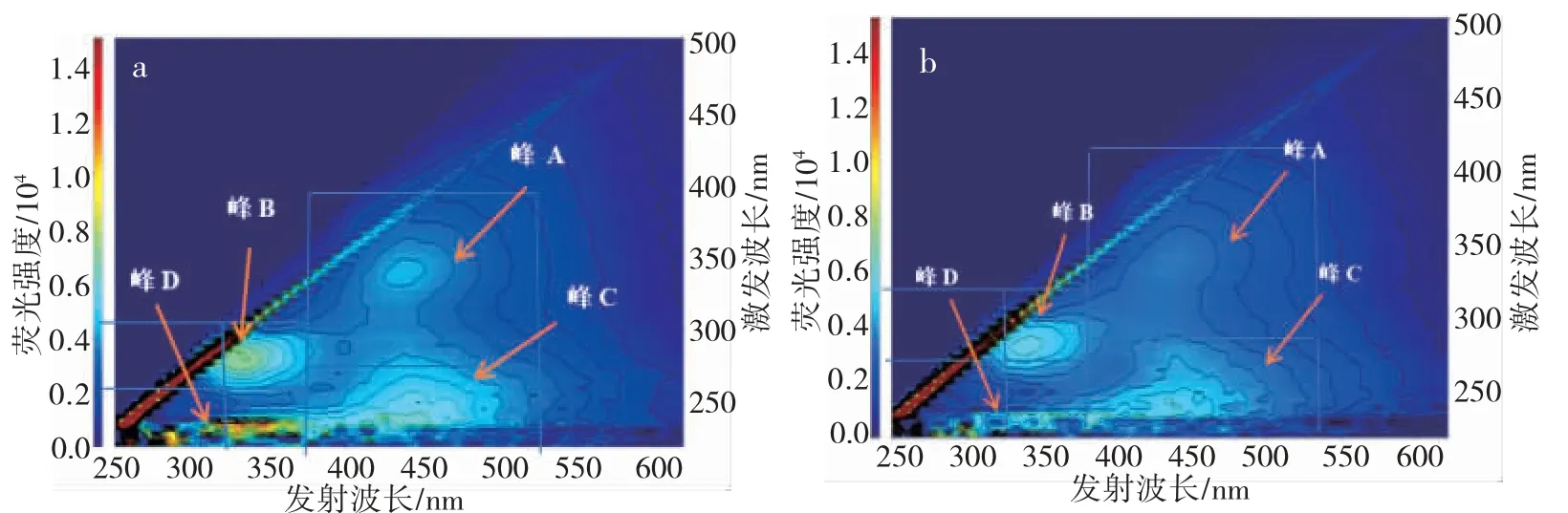
图6 LB-EPS 吸附铬前后三维荧光光谱图(a:吸附前;b:吸附后)Fig. 6 3D-EEM spectra of LB-EPS after chromium adsorption (a: before; b: after)
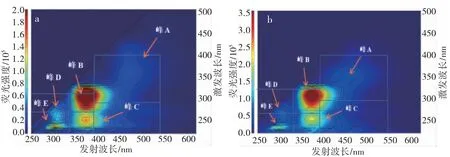
图7 TB-EPS 吸附铬前后三维荧光光谱图(a:吸附前;b:吸附后)Fig.7 3D-EEM spectra of TB-EPS after chromium adsorption (a: before; b: after)
LB-EPS 和TBEPS 中组份及含量存在显著差异,对比吸附铬前后图可知,LB-EPS 中 峰 A 类腐殖酸区域、峰B 类色氨酸区域和峰C 类富里酸区域,反应后荧光强度明显降低;TB-EPS 中峰A类腐殖酸区域、峰C 类富里酸区域、峰D 类酪氨酸区域和峰E 类色氨酸区域,反应后荧光强度明显降低,说明铬与EPS 中的物质发生相互作用,导致荧光猝灭现象。
2.3.2 红外光谱分析
由图 8 (a) 可知,LB-EPS 吸附 Cr(III)后位于 3442 cm-1、1631 cm-1酰胺Ⅰ峰发生偏移。说明铬与多糖表面的O-H 键发生络合,大部分羟基及酰胺基团与Cr(III)发生络合反应。图8(b)可知,TB-EPS 吸附Cr(III)后位于1631 cm-1处和1127 cm-1处峰发生偏移,位于3439 cm-1羟基吸收峰减弱,TB-EPS 上的 -OH、C-O-C 及C-OH 代表的多糖核酸类、-COOH 为Cr(III)提供了吸附位点。吸附后,谱图新增处于1589 cm-1酰胺Ⅱ谱峰,处于1410 cm-1含羧基碳氢化合物谱峰,这可能是由于微生物在重金属铬的胁迫下产生含蛋白质和羧基碳氢化合物[22-23]。进一步说明活性污泥与Cr(III)不是只发生离子交换吸附而是Cr(III)与EPS发生配位作用。
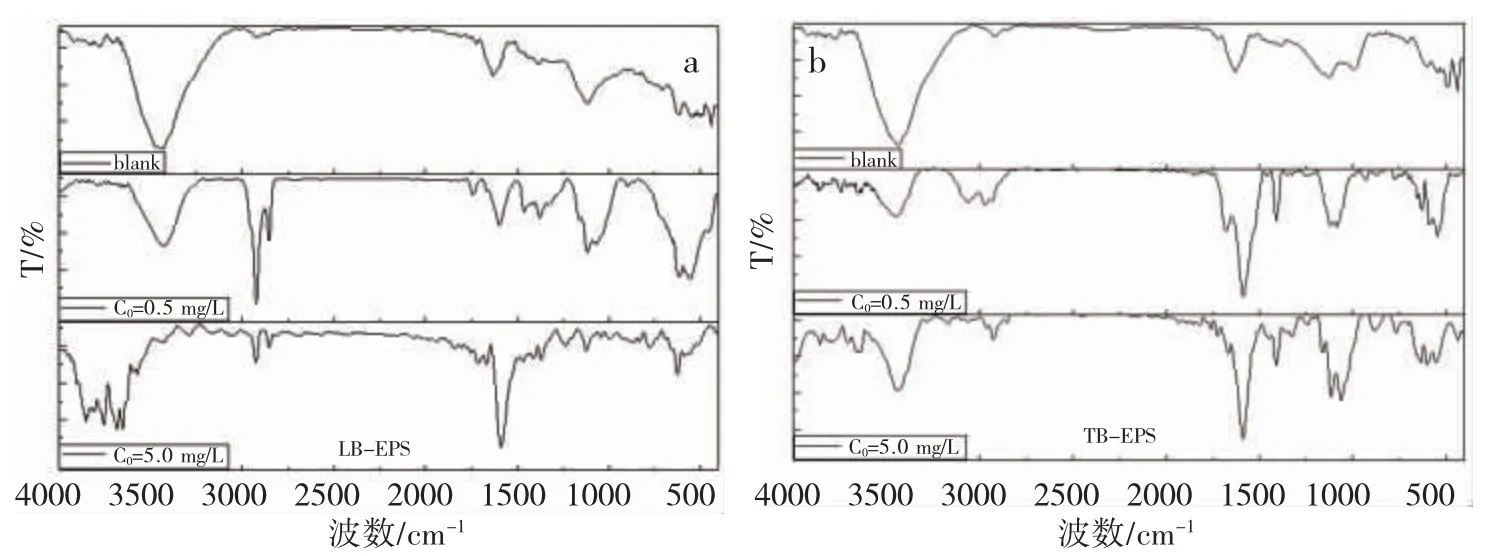
图8 EPS 吸附不同浓度重金属铬后的 FI-TR 图(a:LB-EPS;b:TB-EPS)Fig. 8 FI-TR spectra of EPS after adsorption of different concentrations of heavy metal chromium(a: LB-EPS; b: TB-EPS)
3 结论
(1)制革含铬有机废水经生化处理前后上清液有机络合态铬的分子粒径集中分布在大于450 nm、2~3 nm 和小于 2 nm 三个区域,小于 8 nm 的区域有机物去除率达77.71%,小于5 nm 的区域Cr(III)去除率达68.55%,活性污泥将不溶性含Cr(III)物质完全吸附,络合态铬部分被吸附,吸附后残留少量有机络合态铬。
(2)好氧活性污泥在2 h 内对COD 和Cr(III)同步吸附,活性污泥多次吸附后COD 去除率不足15%,Cr(III)去除率达92.5%,随吸附次数增加,活性污泥对COD 的吸附基本达到饱和,对Cr(III)可以持续吸附。
(3)附着于污泥表面的Cr(III)逐步进入EPS,少量进入微生物内部。低浓度Cr(III)促进微生物细胞分泌EPS,各结构层EPS 蛋白、多糖含量均升高;高浓度Cr(III)抑制微生物细胞分泌EPS,多糖含量显著减少。铬较多分布在TB-EPS 和LB-EPS 中且含量相当。结合光谱分析结果,TB-EPS 上的-OH、C-O-C 及C-OH 代表的多糖核酸类、-COOH 为Cr(III)提供吸附位点。Cr(III)刺激活性污泥中微生物产生酰胺Ⅱ类蛋白质,使EPS 的蛋白质含量增长,活性污泥与Cr(III)不是只发生离子交换吸附而是Cr(III)与EPS 发生配位作用。
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05 07:20:38
上海公路(2019年2期)2019-10-08 09:05:32
中央民族大学学报(自然科学版)(2018年3期)2018-11-09 01:16:42
中央民族大学学报(自然科学版)(2018年3期)2018-01-12 06:03:03
上海公路(2017年1期)2017-07-21 13:38:33
计算机测量与控制(2017年6期)2017-07-01 16:24:28
长江蔬菜(2016年10期)2016-12-01 03:05:36
中国铁道科学(2016年2期)2016-03-30 02:06:59
交通科学与工程(2015年1期)2015-12-23 11:08:07
中国工程咨询(2015年7期)2015-02-14 05:55:52
