
南京雾过程对大气气溶胶谱分布及化学组成的影响
2022-12-20 01:30张思蕊樊曙先胡春阳张鸿伟朱丹丹葛攀延
中国环境科学 2022年11期
张思蕊,樊曙先,王 元,胡春阳,张鸿伟,朱丹丹,葛攀延,2
南京雾过程对大气气溶胶谱分布及化学组成的影响
张思蕊1,2,3,樊曙先1,2*,王 元1,4,胡春阳1,5,张鸿伟1,朱丹丹1,葛攀延1,2
(1.南京信息工程大学,气象灾害预报预警与评估协同创新中心,中国气象局气溶胶与云降水重点开放实验室,江苏 南京 210044;2.南京信息工程大学大气物理学院,江苏 南京 210044;3.上饶市气象局,江西 上饶 334000;4.兰州大学,西部生态安全省部共建协同创新中心,甘肃 兰州 730000;5.中国人民解放军94582部队,河南 确山 463217)
为探究南京地区雾过程对气溶胶粒子化学组成和尺度分布的影响,在2017年冬季的雾观测中平行收集了3级分档雾水和分粒径气溶胶样品,并对雾微物理量与气溶胶谱分布、3级分档雾水与雾前、雾中、雾后分粒径气溶胶化学组成对比分析.结果表明,2017年冬季南京第1次雾过程的雾滴液态水含量随粒径分布为不对称“V”型,最低值位于7μm处,第2次雾过程的雾滴液态水含量随粒径分布为3峰型,峰值分别位于5,15,21.5μm处.在雾形成、发展阶段,粒径<0.33μm的气溶胶质量浓度降低,粒径0.38μm气溶胶质量浓度升高,雾成熟阶段,气溶胶粒子质量浓度在全粒径段均达到最低,粒径0.38μm的气溶胶质量浓度大幅降低,与雾前相比,雾后气溶胶质量浓度峰值向大粒径方向移动.雾前,气溶胶水溶性离子组分富集在粒径<0.43μm的小粒子中,随着雾过程进行,成核作用和吸湿增长使得水溶性离子向较大粒径段富集.雾中新生成的气溶胶随着雾滴的蒸发被释放,导致雾后NO3-、SO42-和NH4+浓度升高.较小粒径的气溶胶中和率更高,雾形成初期的新生雾滴酸性较强,随着雾过程的进行逐渐中和,雾水pH值逐渐升高.
雾微物理结构;化学组分;雾和气溶胶;粒径分布
雾是由空气和水凝物构成的悬浮在大气边界层内的可见聚合体[1-2],可被视为大气气溶胶和气态污染物的物理化学处理器[3].气溶胶粒子中的一部分作为云凝结核(CCN)参与雾过程,其化学组成和尺度分布与雾过程密切相关[4-5].国外开展了大量雾的观测试验来研究雾的宏微观特征[6-18], 20世纪80年代以来国内也相继对城市雾、高山雾、海雾进行了外场观测研究[19-26].研究发现,在液态水含量(LWC)不变的前提下,气溶胶数浓度的增加会导致雾滴数浓度增加[27-28].雾形成之后,大气中的可溶性气体和气溶胶粒子中的可溶性成分可通过液相化学反应进入雾滴,后续还有间隙气溶胶粒子(在雾形成过程中未活化的粒子)与雾滴的碰并以及雾滴之间的碰并过程也将改变气溶胶的化学组成分布[29].在雾形成和发展过程中,雾滴谱分布也受到气溶胶粒子尺度分布的影响[2].雾通过清除、沉降和液相化学反应来改变大气气溶胶的化学组分,而这些作用在具体雾过程中是不同的,与雾滴的大小和含水量、气溶胶的化学组成、吸湿性等因素都有关系.相关研究表明[30],雾对PM10和PM2.5的清除效率分别为55.7%和52%,持续时间越长的雾过程对气溶胶粒子的清除效率越高.因此在对雾进行研究时应将雾与大气气溶胶相结合.
近年来在南京北郊进行了一系列3级分档雾水化学特性的研究[31-33],并评估了南京和庐山3级分档雾水化学特性的差异[34],也有对南京地区气溶胶粒子化学组成随粒径分布和季节变化的研究[35-38],这些研究主要集中于雾水或气溶胶粒子的采样分析,未将二者作为一个完整体系来研究雾过程对气溶胶粒子化学组成和尺度分布的影响.本文在2017年冬季南京的雾观测中平行收集了3级分档雾水和气溶胶样品,基于雾微物理量和气溶胶粒子谱分布,结合3级分档雾水和分粒径气溶胶的化学组成,对2017年冬季南京雾过程中雾滴的数浓度、液态水含量等微物理量和雾前、雾中与雾后气溶胶粒子质量谱分布对比分析,对3级分档雾水和雾前、雾中与雾后分粒径气溶胶化学组分对比分析,以加深对该地区雾与气溶胶相互作用的认识.
1 观测与方法
1.1 观测地点与仪器
南京地处长江中下游,濒江近海,冬季水汽丰富,空气相对湿度大,常出现浓雾天气.大气中化学组分受人为污染源影响较大,具有经济高速发展的区域大气复合污染基本特征.观测地点设在南京市北郊南京信息工程大学校园内(118°42E,32°12N,22m.),局地主要受工厂排放及交通源排放的影响,且观测期间附近有道路和建筑施工.

表1 观测仪器
观测所用仪器见表1. FM-100型雾滴谱仪通过气流速度来计算进气体积,基于光散射原理,测量通过激光束的雾滴产生的散射光,根据不同尺寸的雾滴对激光的散射强度不同,将雾滴进行分档和计数[39].WPS-1000XP型宽范围粒径谱仪的基本原理是测定每一粒子通过两束近距离激光束的飞行时间,以此来换算气溶胶粒子的空气动力学直径,进气口前接有TSI3062干燥管,以保证仪器内部气体相对湿度低于40%,测量结果均为被干燥后的气溶胶[40].主动式3级分档雾水采集器(three-stage CASCC)将雾滴与空气的混合物吸入并撞击在不同规格的特氟龙管子上,各级粒径雾滴被分离并凝结,由于重力的作用沿倾斜的特氟龙管子汇集并通过细软管流入聚乙烯收集瓶,根据粒径大小将雾滴分为3级,切割率为50%,流速为1500L/min[41-42]. FA-3型气溶胶粒度分布采样器为9级撞击采样器,其原理是利用惯性碰撞使悬浮的气溶胶粒子通过进气口后撞击在滤膜上,根据粒子的速度惯性和动力学吸引力原理来实现对不同粒径气溶胶粒子的分离,能够把不同粒径的气溶胶粒子分别采集到各级滤膜上,流速为28.3L/min.
1.2 观测与样品采集
观测前对雾滴谱仪和宽范围粒径谱仪进行标定和流量校准.观测期间对雾滴谱和气溶胶粒子谱进行连续测量,观测结束后根据能见度和气象资料筛选数据.根据能见度选取2017年12月30日0:00~9:00和30日14:00~31日12:00的雾滴谱和气溶胶粒子谱数据,以及雾形成前和雾消散后24h的气溶胶粒子谱数据.
观测期间使用FA-3型气溶胶粒度分布采样器和特氟龙滤膜采集分粒径气溶胶样品.采样前用洁净的镊子将Teflon滤膜放置在各级圆盘中,调整流量后开机采样,同时记录采样起、止日期.气溶胶采样器的流速远小于雾水采集器,不足以抽取雾滴进入采样器,采样后的滤膜仍是干燥的,另架设有挡板遮挡雨水.采样时间为每天11:00~次日10:30,采集时长为23.5h.采样后滤膜用锡纸包裹置于冰箱冷藏待后续实验分析. 本文选取了雾前(1组)、雾中(3组)、雾后(1组)分粒径气溶胶样品.
当能见度低于1km时打开主动式3级分档雾水采集器,为防止雨水渗入以及避免风将其他大颗粒杂质吹入采样器,用塑料布和保鲜膜将采集器包裹起来.3级分档雾水的采样时长首先应尽量保证采样体积足够实验室化学分析,其次考虑雾的生消.采样器和收集瓶均在使用前用电阻率为18.2MΩ/cm的超纯水浸泡冲洗3次并自然风干.雾过程结束后关闭雾水采样器并用纯水再次冲洗采样器.共采集到三级分档雾水样品11个.雾水样品于现场立即测量pH值和电导率(EC)后装入收集瓶用锡纸包裹置于冰箱冷藏待实验室分析水溶性离子.
1.3 样品分析
采样前后均将特氟龙滤膜置于干燥皿恒温恒湿处理后用微量天平称重,采样前后滤膜的重量差即为所采集的分粒径气溶胶重量.气溶胶滤膜样品用洁净的不锈钢刀片截取1/4置于15mL PET瓶中,加10mL超纯水,机械震荡1h后再超声震荡0.5h,静置24h后抽取7mL上清液经0.22μm水系过滤头过滤以备分析. 将雾水样品抽取7mL上清液并用0.22μm的水系过滤头过滤以备分析.
本研究使用ICS 5000+高压离子色谱仪(美国 Thermo Scientific Dionex)测定水溶性离子浓度,测量的水溶性离子包括: Na+、NH4+、K+、Mg2+、Ca2+、Cl-、NO2-、NO3-、SO42-、F-、Br-、C2O42-,具体分析条件和数据质量控制详见朱丹丹等[32].
1.4 数据处理
1.4.1 浓度数据处理 文中所用到的离子浓度均为当量浓度(μeq/L).将离子色谱仪测得离子浓度换算为当量浓度的计算公式为:
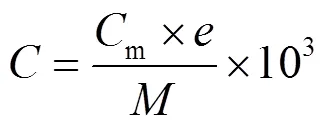
式中:m为各离子组分的质量浓度,mg/L;为各离子的电荷数;为各离子物质的量,g/mol.
1.4.2 雾微物理量计算 在处理雾滴谱数据时,雾滴数浓度f(cm-3)、液态水含量LWC(g/m3)的计算公式分别为:
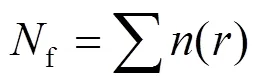
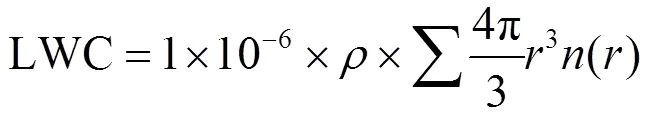
式中:为每档雾滴数浓度,cm-3;为雾滴平均半径,μm;为纯水密度,1.0g/cm3.
1.4.3 气溶胶粒子谱计算 在处理气溶胶粒子谱数据时,气溶胶粒子数浓度a(cm-3)、气溶胶粒子质量浓度d/dlog(μg/m3)的计算公式分别为:
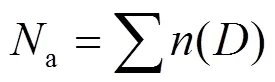
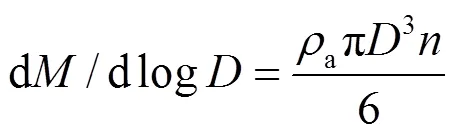
式中:为每个通道的气溶胶粒子数,cm-3;为气溶胶粒子平均粒径,μm;a为气溶胶粒子密度,μg/m3.
1.4.4 气溶胶样品质量浓度和离子浓度的计算 滤膜上所采集的气溶胶质量浓度a(μg/m3)和气溶胶样品中的离子浓度C(μg/m3)用下式计算:

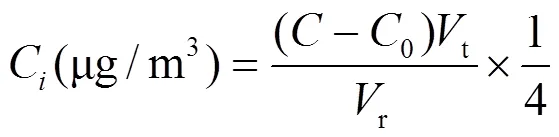
式中:为气溶胶样品液中离子浓度,mg/L;0为空白溶液中离子浓度,mg/L;t为气溶胶样品液体积,为10mL;r为标准状态下气溶胶粒度分布采样器采样体积,m3.
2 结果与分析
2.1 雾过程中雾微物理量和气溶胶粒子谱分布的时间演变
由图1可见,此次雾过程的形成机制是持续输送的冷平流伴随着长波辐射冷却,12月29日下午的毛毛雨使近地面维持较高相对湿度,稳定的大气层结有利于雾形成.12月30日0:00起,雾开始形成(N>1cm-3且LWC>0.001g/m3),地面能见度降低至1km,其后在1km上下持续浮动,《地面气象观测规范》[43]中,将雾定义为大量小水滴悬浮在低层大气中使地面水平能见度小于1km的天气现象,将水平能见度在1.0~10.0km的雾定为轻雾.与霾相比,雾的相对湿度更高,接近100%.0.1~0.3μm粒径段气溶胶在30日0:00具有较高数浓度,同时,PM2.5和PM2.5~10质量浓度也较高.在高过饱和度条件下,CCN浓度与气溶胶数浓度具有很好的正相关性[44].此次雾过程中爱根核模态和积聚模态的气溶胶粒子是CCN的主要贡献者. 随着能见度的持续下降,0.3μm以下粒径段气溶胶被不断活化,数浓度持续降低,伴随着相对湿度的逐渐升高,雾滴数浓度和液态水含量升高,雾滴逐渐增大,雾滴数浓度与能见度成负相关.到1:00,雾滴数浓度由100cm-3量级增至101cm-3量级,雾谱拓宽,液态水含量也增加.随着雾过程的进行,气溶胶数浓度、大气PM2.5和PM2.5~10质量浓度均降低,这意味着雾对气溶胶的有效清除.7:30之后,雾滴数浓度下降,近地面能见度逐渐回升,PM2.5质量浓度升高,但PM2.5~10质量浓度并没有回升,这是由于新生成的气溶胶粒子集中在粒径<2.5μm以下,而粗颗粒物受到雾滴的沉降作用被沉降至地面.气溶胶粒子谱可以更清晰的显示在0.01~0.3μm粒径段有数浓度的急剧升高,这也意味着新生成的气溶胶粒子尺寸较小,且新生成的气溶胶粒子在高湿条件下进一步吸湿增长.由于相对湿度高且气溶胶数浓度偏低,此次雾过程的主要特征为:雾滴数浓度低、雾滴粒径偏小且液态水含量高,并且雾滴液态水含量在雾形成阶段达到最高.
由图2可见,30日14:00~18:00雾形成阶段,近地面能见度在1km上下波动,气溶胶数浓度升高以粒径<0.1μm的气溶胶粒子为主,相较于第一次雾过程,此次雾过程CCN的贡献者主要是爱根核模态气溶胶粒子,气溶胶粒子粒径偏小,数浓度偏高. PM2.5和PM2.5~10质量浓度峰值更高.到18:00,地表长波辐射冷却使雾得以发展,近地面能见度降低至500m左右,0.03~0.06μm粒径段的气溶胶粒子数浓度超过500cm-3.到19:00,气溶胶粒子吸湿碰并增长并且数浓度升高,粒径0.04~0.1μm的气溶胶粒子数浓度超过600cm-3,其中0.06μm段的粒子数浓度最大达到700cm-3,在此阶段,雾滴数浓度维持在100cm-3量级,且雾滴谱很窄,主要为5μm以下的小雾滴,雾滴液态水含量约在10-5g/m3量级.随着雾的发展,雾滴数浓度和尺寸均有所增加,雾滴谱拓宽,雾滴液态水含量升高到10-4g/m3量级,气溶胶粒子尺寸继续增大,粒径<0.02μm的纳米级气溶胶粒子数浓度降低,PM2.5质量浓度持续下降,这表明了雾发展阶段气溶胶粒子持续吸湿增长.31日3:00之后,小雾滴的数浓度和液态水含量均有明显增加,同时,粒径<0.05μm的气溶胶粒子数浓度下降,表明此粒径段的气溶胶粒子对小雾滴的生成贡献最大.31日5:00~11:00,雾成熟阶段,近地面能见度骤降至50m以下,雾出现爆发性增长,雾滴数浓度骤增2个量级,雾滴尺寸骤增2倍,雾滴谱拓宽明显,同时雾滴液态水含量增加了3个量级.此时>0.2μm粒径段气溶胶粒子数浓度有明显降低,表明积聚模态的气溶胶粒子对雾滴生成的贡献更大.其后,0.02~0.05μm粒径段气溶胶粒子数浓度上升,至9:00~10:00,气溶胶粒子数浓度在0.02~ 0.05μm粒径段达到峰值,表明此时新生成了纳米级气溶胶粒子.之后,新生成的气溶胶粒子吸湿增长,气溶胶粒子谱拓宽,由于气溶胶粒子之间争夺水汽,雾滴液态水含量下降,雾滴发展受到限制,雾滴数浓度和尺寸均减小.11:00之后,由于太阳短波辐射的增强,雾逐渐消散,能见度上升至1km之上.此次雾过程具有爆发性增长的特征,雾滴数浓度高、粒径偏大,同时生成了大量纳米级气溶胶粒子.
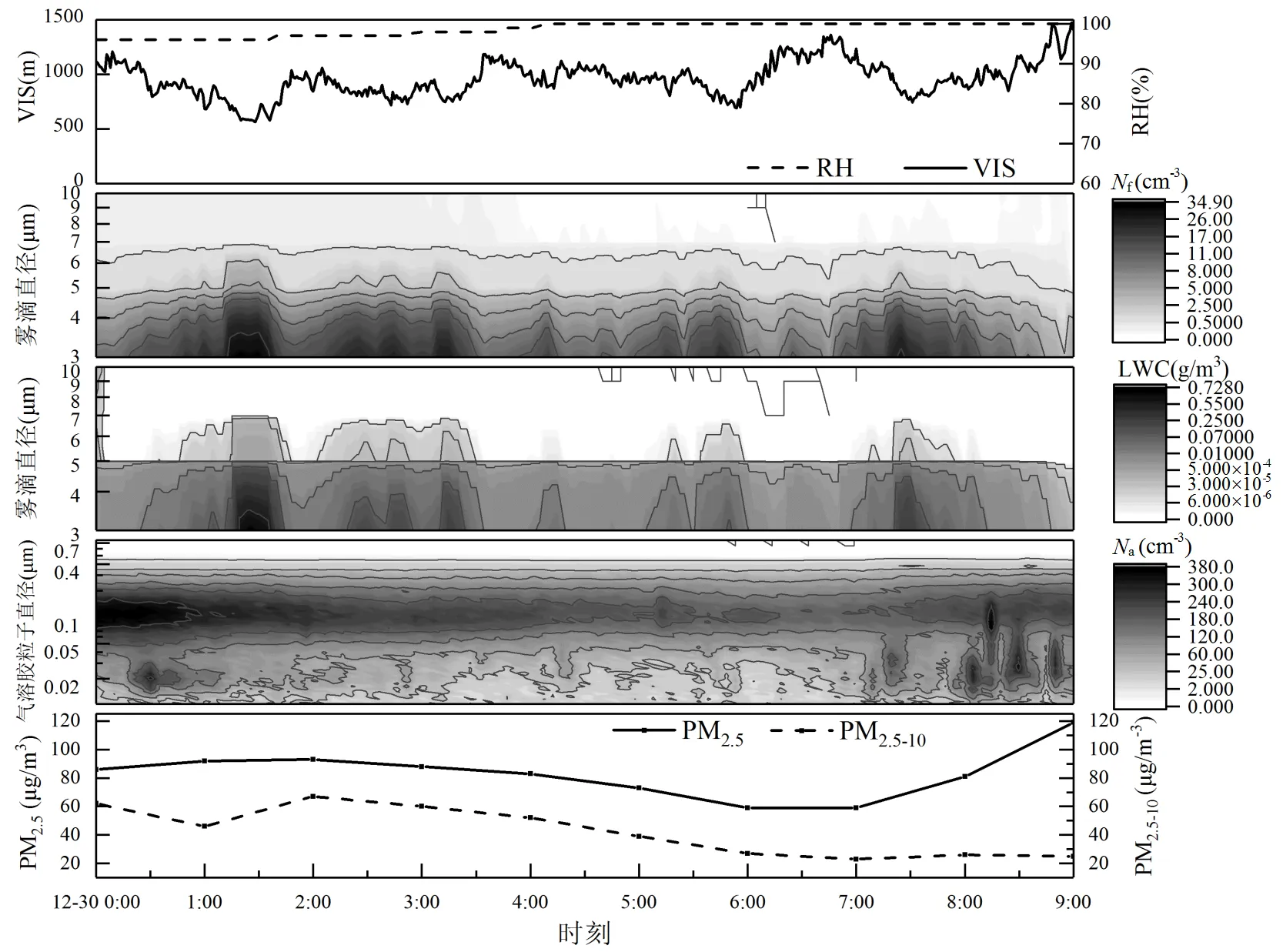
图1 2017年12月30日0:00~9:00地面能见度(VIS)、湿度(RH)、雾滴数浓度(Nf)、雾滴液态水含量(LWC)、气溶胶粒子数浓度(Na)、PM2.5和PM2.5~10质量浓度的时间序列

图2 2017年12月30日14:00~31日12:00地面能见度(VIS)、湿度(RH)、雾滴数浓度(Nf)、雾滴液态水含量(LWC)、气溶胶粒子数浓度(Na)、PM2.5和PM2.5~10质量浓度的时间序列
2.2 雾滴液态水含量(LWC)和气溶胶粒子质量谱随粒径的分布特征
2.2.1 雾滴液态水含量随粒径的分布 雾滴液态水含量的粒径分布特征与雾的生消过程、气象要素、湍流活动、地理环境等多种因素相关[39].Niu等[45]对南京2006~2009年冬季雾滴谱的分析发现,雾滴液态水含量随雾滴粒径分布的峰值位于5,13和21.5μm处,2017年冬季南京第1次雾过程在轻雾与雾之间转换,雾滴液态水含量峰值位于3μm处,次峰位于49μm处,雾滴液态水含量随粒径的分布为不对称“V”型,最低值位于7μm处(图3a).此次雾过程的雾滴液态水含量主要集中在3~5μm小雾滴.而第2次强浓雾过程的雾滴液态水含量随粒径的分布与Niu等[45]的研究结果类似,为3峰型,峰值分别位于5,15,21.5μm处(图3b).
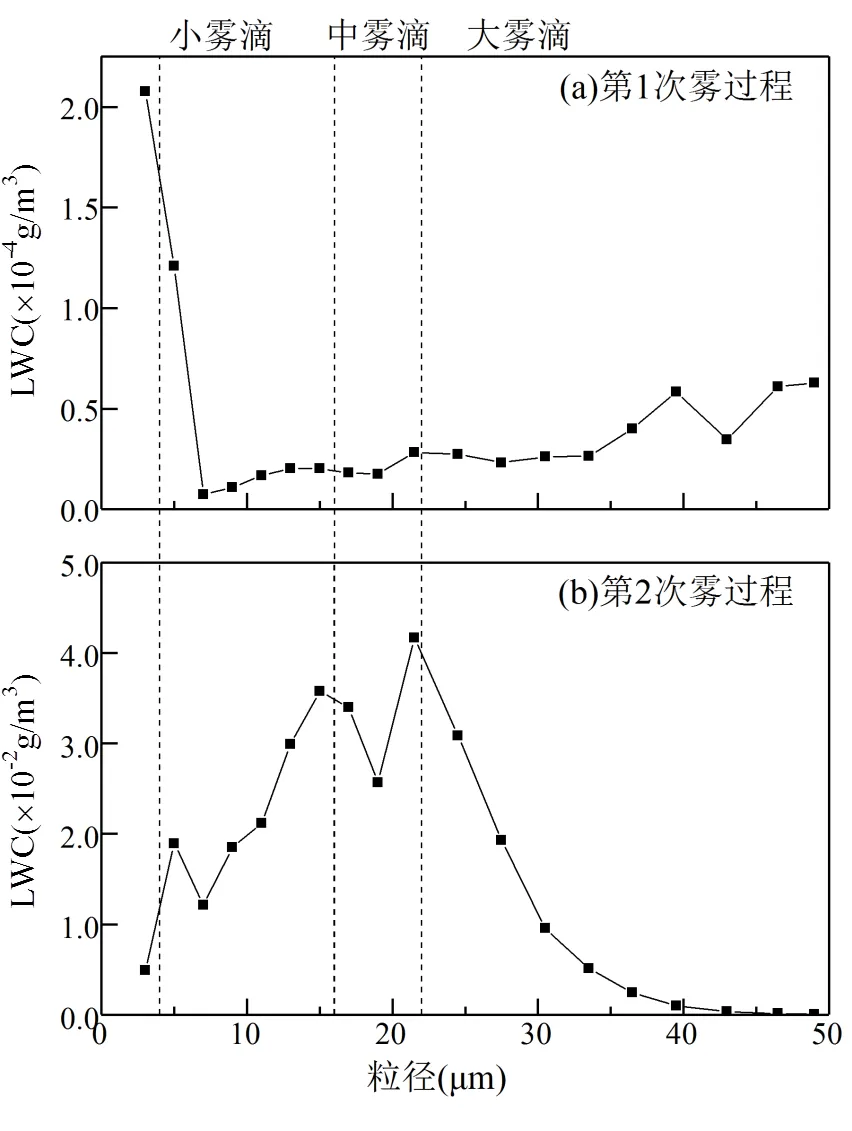
图3 2017年冬季南京两次雾过程的雾滴液态水含量(LWC)随雾滴粒径变化
雾滴液态水含量的粒径分布从本质上决定着本文用three-stage CASCC采集到的三级分档雾水量,为进一步研究三级分档雾水样品分布的不均匀特征,将雾滴液态水含量随粒径的分布按照雾水样品的采集时间分析.由图4可见,雾滴液态水含量在相应的雾水样品三级切割直径区间内与本文所采集到的雾水样品量具有一致性,当雾滴液态水含量在4~16μm粒径段较高时,4~16μm粒径段的雾水样品量也较多,证明观测结果具有可靠性.雾水样品的采集量不仅与能见度有关,还与液态水含量和雾滴尺度有关,第一次雾过程虽然近地面能见度平均值(960m)比第二次雾过程(484m)高,雾滴数浓度平均值(16.92cm-3)比第二次雾过程(515.84cm-3)低,但能够采集到雾水,是由于雾滴液态水含量峰值(0.73g/ m3)比第二次雾过程(0.23g/m3)高.
2.2.2 雾前、雾中与雾后气溶胶粒子质量谱随粒径的分布 雾与气溶胶之间存在着复杂的双重关系,雾滴增长受气溶胶物理化学特性的影响,同时雾过程能改变气溶胶的粒径分布[46].Hussein等[47]将气溶胶根据粒径分为核模态(3~25nm)、爱根核模态(25~100nm)、积聚模态(100~1000nm)和粗粒子模态(>1µm).根据柯拉理论,>300nm的粒子比较小的粒子更容易激活,特别是在典型的低过饱和度条件下.在气溶胶粒子数浓度的时间序列(见图1、2)中已经观察到,雾形成阶段,气溶胶数浓度较高,随着雾的发展,气溶胶数浓度下降,雾即将消散时气溶胶数浓度又升高.为研究雾过程对气溶胶粒子谱分布的影响,分析雾前、雾中与雾后气溶胶粒子质量浓度谱分布(图5a)和数浓度谱分布(图5c)变化.雾前气溶胶粒子质量谱呈双峰分布,主峰约在0.28μm处,0.38μm处也有一小的次峰.雾中,粒径<0.33μm的气溶胶粒子质量浓度逐渐下降,而粒径>0.36μm的气溶胶粒子质量浓度上升,也就是说,与雾前相比气溶胶粒子质量浓度的主峰峰值降低,而次峰峰值升高,图5c可见,粒径<0.3μm的气溶胶粒子数浓度显著下降,0.38μm粒径段的气溶胶粒子数浓度略增,由于雾清除作用,核模态和爱根核模态气溶胶粒子活化并被雾滴不断吸附.到雾后,由于雾滴蒸发释放出雾滴中生成的新粒子,核模态和爱根核模态气溶胶粒子数浓度大幅升高,质量浓度谱分布中,0.38μm粒径段成为气溶胶粒子质量浓度主峰,此粒径段质量浓度的增加主要是由数浓度增加贡献.而<0.3μm粒径段的气溶胶质量浓度比雾中更低,虽然<0.08μm粒径段的气溶胶数浓度升高,但由于其粒径小,对质量浓度贡献小.与雾前相比,气溶胶质量浓度峰值出现了向大粒径方向移动的现象,这与樊曙先等[48]2007年的研究结果一致.而数浓度峰值向小粒径方向移动,雾过程改变了气溶胶的粒径分布特征.

图4 2017年冬季南京三级分档雾水样品采集时段雾滴液态水含量(LWC)随雾滴粒径的变化
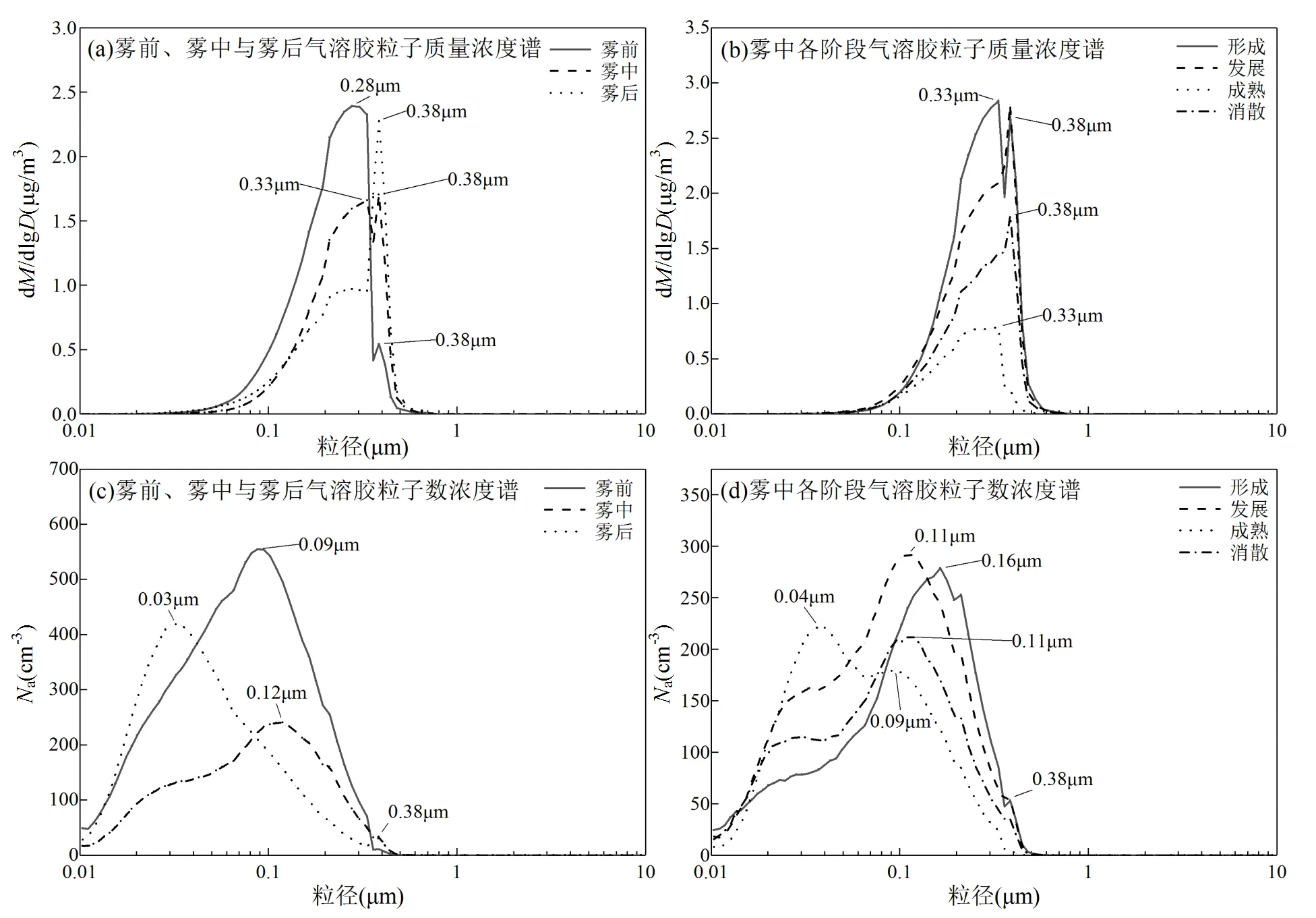
图5 2017年冬季南京雾前、雾中与雾后和雾中各阶段气溶胶粒子质量浓度谱分布和数浓度谱分布
碰并过程主要发生在雾的发展和成熟阶段[49].进一步研究雾过程中各阶段气溶胶粒子质量浓度谱分布(图5b)和数浓度谱分布(图5d)变化,在雾的形成阶段,气溶胶粒子(主要是核模态和爱根核模态)数浓度显著下降,这主要是由于气溶胶粒子的核化过程,此时质量浓度谱分布呈双峰型,峰值分别位于0.33和0.38μm处.到雾发展阶段,爱根核模态气溶胶粒子数浓度上升,积聚模态气溶胶粒子数浓度下降,粒径<0.38μm的气溶胶粒子质量浓度持续下降, 0.38μm粒径段气溶胶粒子质量浓度和数浓度略微升高.雾的成熟阶段,核模态气溶胶粒子数浓度增加,有纳米级气溶胶粒子生成,而爱根核模态和积聚模态气溶胶粒子数浓度降低,气溶胶粒子数浓度峰值向小粒径方向移动,雾的清除作用使得气溶胶粒子质量浓度在全粒径段均达到最低水平,此时气溶胶粒子质量浓度谱分布呈单峰型,峰值约0.33μm.到雾的消散阶段,雾滴蒸发释放出气溶胶粒子,气溶胶粒子数浓度峰值又重新向较大粒径方向移动,粒径<0.07μm的气溶胶数浓度下降,粒径>0.07μm的气溶胶数浓度上升,这也导致气溶胶质量浓度又升高,此时气溶胶粒子的质量浓度峰值已经移动0.38μm粒径段.因此,雾过程不同阶段中,由于核化、凝结、碰并和沉降等物理过程同时存在但主导地位不同,气溶胶粒子质量浓度谱分布呈现出不同的变化特征.
2.3 雾前、雾中与雾后分粒径气溶胶和3级分档雾水的化学特征
2.3.1 雾前、雾中与雾后分粒径气溶胶质量浓度及水溶性离子的分布特征 气溶胶化学组分的粒径分布是气溶胶的重要性质之一[50].气溶胶化学组分对其吸湿性有重要影响,从而影响雾的发生发展[51],作为雾滴的凝结核,雾前气溶胶的化学组分从一定程度上决定了雾滴的初始化学组成,而随后雾的凝结、沉降、非均相化学反应、液相化学反应和蒸发等过程又影响气溶胶中化学组分的分布[52].为深入了解气溶胶化学组成分布和雾滴化学组成分布的相互影响,分析雾前、雾中与雾后分粒径气溶胶的质量浓度和化学组成变化.图6(a)表明,气溶胶质量浓度由高到低为:雾中>雾前>雾后,总水溶性离子浓度由高到低也为:雾中>雾前>雾后.由于FA-3气溶胶粒度分布采样器是将空气吸入使得气溶胶粒子撞击在滤膜上,雾中采集到的气溶胶包括间隙气溶胶和雾滴蒸发后释放出的核气溶胶,因此雾中气溶胶质量浓度和总水溶性离子浓度均为最高.雾中,气溶胶质量浓度和总水溶性离子浓度约为雾前的2倍,雾后,气溶胶质量浓度和总水溶性离子浓度再次降低,并且低于雾前浓度水平.各水溶性离子种类在雾前、雾中与雾后的浓度变化也有所差异,2017年南京气溶胶中最主要的水溶性离子为NO3-、SO42-、NH4+和Cl-(图6b).气溶胶中的主要离子在雾前、雾中与雾后均存在明显差异.雾前,Ca2+的浓度占比很高(14%),到了雾中降低(5%),Ca2+主要存在于粗粒子模态(>1μm)气溶胶,这表明雾滴对较大粒径的气溶胶粒子沉降作用更大.雾中增幅最明显的是NO3-、SO42-和NH4+,雾前和雾中NO3-浓度占比很高,而雾后SO42-的浓度占比升高到约与NO3-相当,这是一个很有趣的现象,气溶胶中NO3-与SO42-浓度比值在雾前、雾中、雾后分别为1.80、1.69、1.09,随着雾过程的进行浓度比值逐渐降低,表明雾对NO3-与SO42-的转化效率是不同的,在相对湿度较高的环境下,更有利于SO42-的生成.有气溶胶中化学组分对活化率的影响研究表明,无机组分比有机组分的吸湿性高,而硝酸盐在无机组分中吸湿性相对较高[53-54].雾前NO3-浓度占比最高,气溶胶吸湿性更强,有利于雾形成,随着雾的发展,NO3-浓度占比下降,气溶胶粒子吸湿性降低,阻碍雾的维持.因此NO3-浓度占比降低而SO42-的浓度占比升高也表明随着雾的发展气溶胶粒子的吸湿性逐渐降低,影响着雾过程的生消.
空气动力学直径在140nm~1.2μm之间的气溶胶受雾过程的影响最大[55].进一步分析雾过程对气溶胶水溶性离子质量浓度和化学组成随粒径分布方面的影响(图7),雾前,气溶胶水溶性离子组分集中在粒径<0.43μm的粒子中,占到总气溶胶的72%.随着雾过程进行,成核作用和吸湿增长使得粒径<0.43μm的气溶胶质量浓度大幅下降,较大粒径段的气溶胶水溶性离子质量浓度均升高,呈现双峰分布,峰值分别位于1.1~2.1μm和5.8~9.0μm处,主峰从<0.43μm移动到1.1~2.1μm处,这进一步证实了作为雾滴凝结核的气溶胶粒子主要在0~0.43μm粒径段内.雾后气溶胶水溶性离子质量浓度峰值又重新回到<0.43μm粒径段,且雾后总水溶性离子质量浓度与雾前相比,除了<0.43μm和9~10μm粒径段有下降,其余粒径段的质量浓度均有上升.NH4+在气溶胶中的占比呈现在小粒子中最高,NO3-占比也呈现在小粒子中更高,NO2-占比在大粒径气溶胶中较高.由于雾的沉降作用,雾后气溶胶中Ca2+浓度与雾前相比有明显下降.雾后NO3-、SO42-和NH4+的浓度占比在各粒径段均有升高,这可能是雾中液相化学反应新生成的气溶胶粒子随着雾滴的蒸发被释放.雾过程改变了水溶性化学组分在气溶胶粒子中的分布特征.

图6 2017年冬季南京雾前、雾中与雾后气溶胶质量浓度、气溶胶中化学组分质量浓度(a)和气溶胶中各水溶性离子组分占比(b)

图7 2017年冬季南京雾前、雾中与雾后分粒径气溶胶总水溶性离子质量浓度和化学组分占比随粒径分布
雾前,NH4+几乎都分布在0~0.43μm气溶胶中, NO3-的浓度占比也在0~0.43μm最高,SO42-在0~ 0.65,9~10μm粒径段的占比最高,这3种组分主要来自于人为源.Cl-集中在0.65~5.8μm的气溶胶中,NO2-呈双峰分布,主要分布在0.43~3.3,5.8~ 10μm粒径段的气溶胶. K+主要分布在0~1.1,3.3~4.7μm的气溶胶中,Ca2+雾前大量分布在0.43~10μm粒径段,Na+雾前在0.65~1.1,3.3~4.7μm的占比较高.
从雾前到雾中,水溶性离子组分均有不同程度的向大粒径气溶胶中移动,这主要是由于雾中气溶胶粒子的吸湿增长. NO3-、SO42-、NH4+、Cl-、Ca2+、Na+和Mg2+的浓度峰值从0~0.43μm移动到1.1~2.1μm,并且在5.8~9.0μm出现第二峰. K+的浓度峰值从0~0.43μm移动到0.43~0.65μm,C2O42-的浓度峰值从0~0.43μm移动到0.43~0.65μm和1.1~2.1μm,呈双峰分布.NO2-和F-浓度呈三峰分布,峰值位于0.43~0.65μm、3.3~4.7μm和9~10μm.在这些水溶性离子组分中,以NO3-的质量浓度变化最大.质量浓度变化为NO3-> SO42-> NH4+> Cl-> Ca2+> K+> C2O42-> NO2-> Na+> Mg2+> F-> Br-.
雾后, NH4+主要分布在0~2.1μm气溶胶中, NO3-和SO42-在各粒径段的占比差别不大,二次离子种类(NH4+、NO3-和SO42-)浓度占比累计在雾后仍然是0~2.1μm细粒子气溶胶中的占比最高. Cl-、Br-和NO2-主要分布在粗粒子模态气溶胶中. Ca2+雾前大量分布在0.43~10μm,Na+雾后在5.8~10μm的占比最高. C2O42-主要分布在0~4.7,5.8~9.0μm气溶胶中. 与雾前相比, NO3-、SO42-、NH4+、Cl-、Br-和NO2-浓度占比也有向较大粒径的粗粒子模态中移动的趋势,这主要是雾中发生的液相反应生成的新粒子由于雾滴的蒸发又释放在气溶胶粒子中.
2.3.2 三级分档雾水和雾前、雾中与雾后分粒径气溶胶酸度随粒径分布特征 酸度在理解雾和气溶胶物理和化学性质方面至关重要,分析三级分档雾水和雾前、雾中与雾后分粒径气溶胶的酸度,有助于理解雾过程对气溶胶化学特性的影响.Lu 等[56]在2006年对南京雾水酸度的研究中已经发现新生雾滴多为酸性,在雾形成后逐渐中和.本文在对2017年南京三级分档雾水pH值的分析也发现类似现象(表2),在第二次雾过程中,随着雾过程的进行,所采集到的三组分档雾水的pH值依次升高,并且呈现酸性在小雾滴中富集的现象.第一次雾过程中的分档雾水pH值略高于第二次雾过程,是由于第一次雾过程在轻雾与雾之间来回转换,轻雾的雾水pH值高于雾[50].而第一次雾过程中第2组雾水中小雾滴的pH值与第1组相比反而降低,由于雾水样品采集的难度与数量限制,无法清晰地呈现其中的规律,但有可能雾水酸度随着雾过程发展而降低这一规律也受到雾微物理结构和气象条件的影响而有所差异.
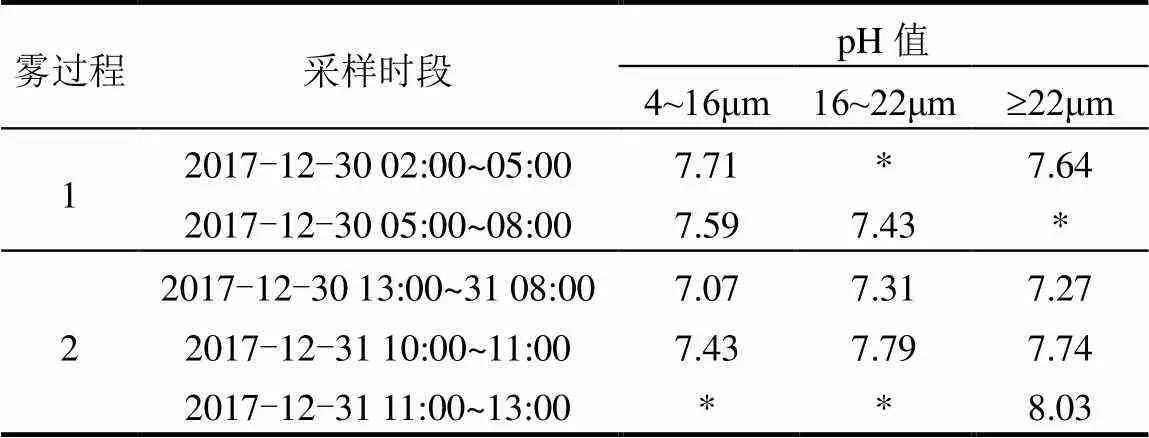
表2 三级分档雾水pH值
注:“*”表示未检测到.
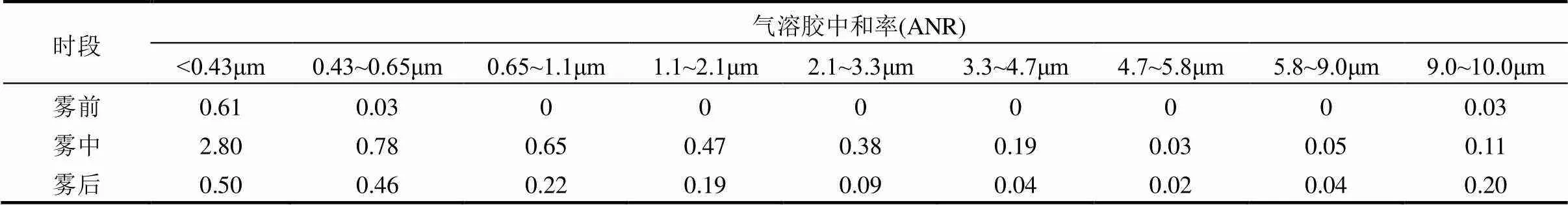
表3 雾前、雾中与雾后分粒径气溶胶中和率
研究表明,雾滴凝结核通常在气溶胶中和状态下形成[29],有雾发生时,间隙气溶胶几乎被中和[57].为了进一步研究雾发生时的中和过程,本文关注到气溶胶的酸度,但其不能直接测量,因此通过计算气溶胶中和率(ANR) 来间接评估气溶胶的酸度[58].
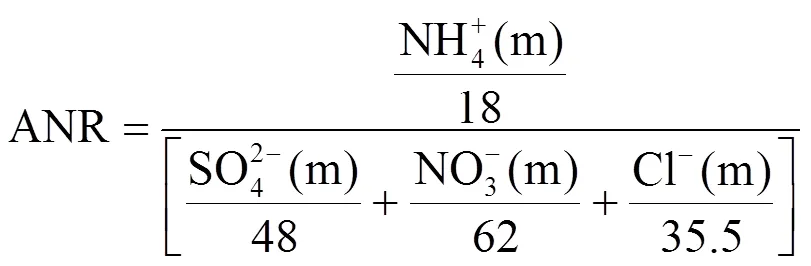
式中:NH4+(m)、SO42-(m)、NO3-(m)、Cl-(m)是样品的测量浓度.
由表3可见,2017年冬季南京气溶胶中和率在雾前很低,随着雾过程的进行,到雾中,气溶胶中和率升高,气溶胶被中和,雾后气溶胶中和率又降低,但雾后的气溶胶中和率高于雾前.在气溶胶中和率随粒径分布方面,<0.43μm的气溶胶在雾前、雾中与雾后的中和率都比其他粒径段高,同前文论述,雾滴的凝结核主要是由此粒径段的气溶胶粒子活化而来.
2.3.3 三级分档雾水和雾中分粒径气溶胶水溶性离子负载 雾滴对可溶性气体和气溶胶粒子的清除以及非均相化学反应过程共同决定了雾水化学组分,在一定程度上可以反映区域大气气溶胶化学组分[59].本文对比了分档雾水和分粒径气溶胶的水溶性离子组分,研究雾中雾水化学组分和气溶胶化学组分的特征.为便于比较雾水和气溶胶中的化学成分浓度,在相同单位(μg/m3)下对其进行评估,计算三级分档雾水中各水溶性离子负载FWL(表4).2017年冬季南京三级分档雾水中的总水溶性离子负载为99.13μg/m3,雾前、雾中与雾后气溶胶总水溶性离子质量浓度分别为39.26, 110.33和29.96μg/m3,这进一步表明,气溶胶粒度分布采样器采集的气溶胶包括:(1)雾中原本存在的间隙气溶胶;(2)雾滴蒸发后释放出的核气溶胶.雾水中的水溶性离子负载从高到低依次为NO3-> SO42-> Ca2+> NH4+> Cl-> K+> Na+> Mg2+,在气溶胶中Ca2+含量低于NH4+,这与Izhar等[30]的研究结果一致,雾水中Ca2+含量更高是由于雾滴沉降作用清除了大粒径的大气气溶胶, Ca2+通常存在于较大的气溶胶中.在气溶胶中, NH4+是主要的碱性离子,其含量很高,并且主要存在于直径3.3μm以下的气溶胶中.雾水中[NO3-]/[SO42-]的平均值为1.29,而气溶胶中[NO3-]/[SO42-]的平均值为1.45,这表明雾滴对二者的氧化和清除效率不同.
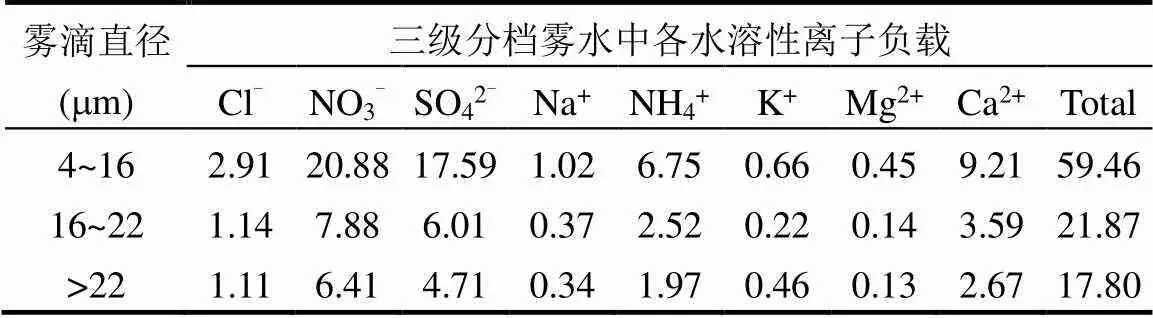
表4 三级分档雾水中各水溶性离子负载(μg/m3)
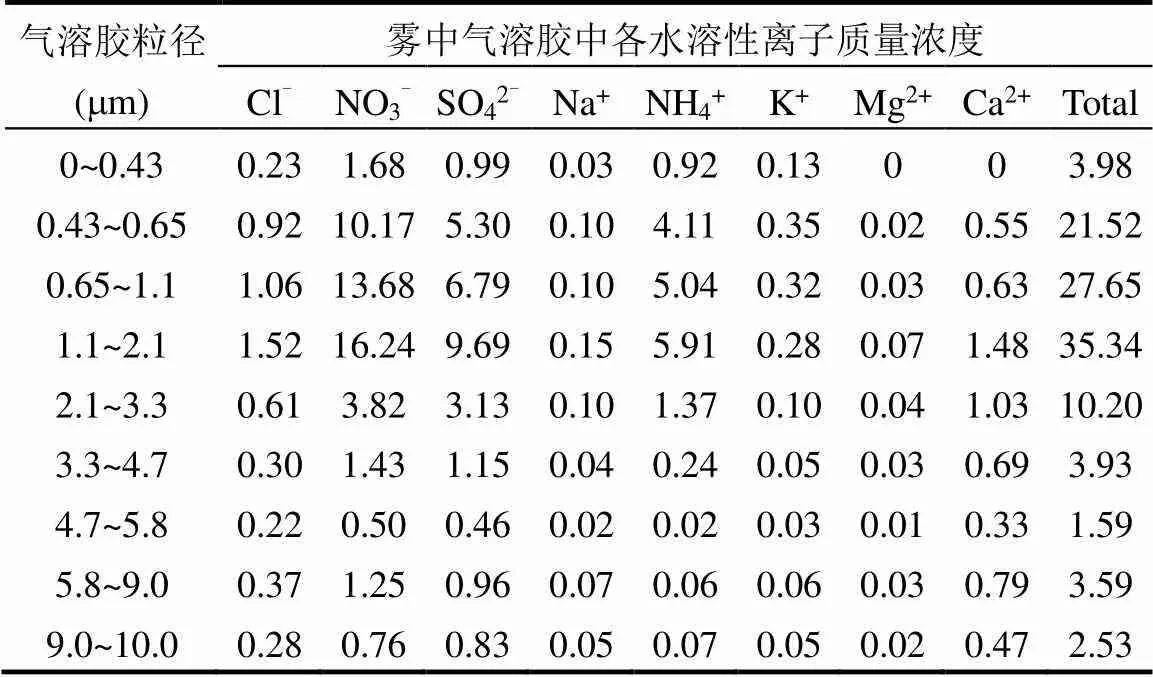
表5 雾中分粒径气溶胶中各水溶性离子质量浓度(μg/m3)
2017年冬季南京三级分档雾水中的总水溶性离子及各水溶性离子负载均呈现在4~16μm小雾滴中最高,这除了与雾水中总离子浓度有关,还与雾微物理结构有关,与雾滴液态水含量的尺度依赖性有关,小雾滴液态水含量高.分粒径气溶胶中水溶性离子质量浓度主要集中在直径3.3μm以下的气溶胶中(表5),0~0.43μm粒径段的气溶胶水溶性离子质量浓度较低,这是由于雾过程中气溶胶粒子吸湿增长,因此水溶性离子组分也向稍大粒径段富集.
3 结论
3.1 2017年冬季在南京共观测到2次雾过程,第一次轻雾过程雾滴数浓度低、雾滴粒径偏小且液态水含量高,雾滴液态水含量随粒径的分布为不对称“V”型,最低值位于7μm处.第2次强浓雾过程存在爆发性增长的特征,且爆发性增长阶段有大量新粒子生成,雾滴液态水含量随粒径的分布为3峰型,峰值分别位于5,15,21.5μm处.
3.2 雾过程改变了气溶胶粒子谱及其化学组成随粒径的分布特征.2017年冬季南京雾前气溶胶粒子质量谱呈双峰分布,主峰位于0.3μm粒径段.与雾前相比,雾后气溶胶质量浓度峰值出现了向大粒径方向移动的现象.雾中,气溶胶质量浓度和总水溶性离子浓度约为雾过程前的2倍,雾消散后,气溶胶质量浓度和总水溶性离子浓度再次降低,并且低于雾过程前浓度水平.
3.3 雾对NO3-与SO42-的转化效率不同,这也与气溶胶的活化率有关.雾前,气溶胶水溶性离子组分富集在粒径<0.43μm的小粒子中,随着雾过程进行,成核作用和吸湿增长使得水溶性离子向较大粒径段的气溶胶中富集.雾中生成的新粒子随着雾的消散而蒸发被释放,使得雾后NO3-、SO42-和NH4+浓度升高.雾滴凝结核通常在气溶胶中和状态下形成,较小粒径的气溶胶中和率更高.
[1] 顾震潮.云雾降水物理基础 [M]. 北京:科学出版社, 1980:1-5.
Gu Z C. Physics basics of cloud precipitation [M]. Beijing: Science Press, 1980:1-5.
[2] 杨 军,陈宝君,银 燕,等.云降水物理学 [M]. 北京:气象出版社, 2011:76-77.
Y J, Chen B J, Yin Y, et al. Physics of clouds and precipitation [M]. Beijing: Meteorological Press, 2011:76-77.
[3] Hoag K J, Collett Jr J L, Pandis S N. The influence of drop size- dependent fog chemistry on aerosol processing by San Joaquin Valley fogs [J].Atmospheric Environment, 1999,33(29):4817-4832.
[4] 张小曳.中国大气气溶胶及其气候效应的研究 [J]. 地球科学进展, 2007,22(1):12-16.
Zhang X H. Study on atmospheric aerosols and their climate effects in China [J]. Advances in Earth Science, 2007,22(1):12-16.
[5] 时宗波,贺克斌,陈雁菊,等.雾过程对北京市大气颗粒物理化特征的影响[J]. 环境科学, 2008,29(3):551-556.
Shi Z B, He K B, Chen Y J, et al. Influence of fog processes on characteristics of individual particles in the urban atmosphere of Beijing [J]. Environmental Science , 2008,29(3):551-556.
[6] Pilié R J, Mack E J, Kocmond W C, et al. The life cycle of valley fog. Part I: Micrometeorological characteristics [J]. Journal of Applied Meteorology, 1975, 14(3):347-363.
[7] Roach W T, Brown R, Caughey S J, et al. The physics of radiation fog: I-a field study [J]. Quarterly Journal Of The Royal Meteorological Society, 1976,102(432):313-33.
[8] Pilié R J, Mack E J, Rogers C W, et al. The formation of marine fog and the development of fog-stratus systems along the California coast [J]. Journal of Applied Meteorology, 1979,18(10):1275-1286.
[9] Lewis J M, Korain D, Redmond K T. Sea fog research in the United Kingdom and United States: A historical essay including outlook [J]. Bulletin of the American Meteorological Society, 2004,85(3):395408.
[10] Gultepe I, Pearson G, Milbrandt J A, et al. The fog remote sensing and modeling field project [J]. Bulletin of the American Meteorological Society, 2009,90(3):341-359.
[11] Fuzzi S, Castillo R A, Jiusto J E, et al. Chemical composition of radiation fog water at Albany, NewYork, and its relationship to fog microphysics [J]. Journal of Geophysical Research, 1984,89(D5): 7159-7164.
[12] Fuzzi S, Laj P, Ricci L, et al. Overview of the Po valley fog experiment 1994 (CHEMDROP) [J]. Contributions to Atmospheric Physics, 1998,71(1):3-19.
[13] Anderson J B, Baumgardner R E, Mohnen V A, et al. Cloud chemistry in the eastern United States, as sampled from three high-elevation sites along the Appalachian Mountains [J]. Atmospheric Environment, 1999,33(30):5105-5114.
[14] Wrzesinsky T, Klemm O. Summertime fog chemistry at a mountainous site in central Europe [J]. Atmospheric Environment, 2000,34(9): 1487-1496.
[15] Collett J L, Sherman D E, Moore K F, et al. Aerosol particle processing and removal by fogs: observations in chemically heterogeneous central California radiation Fogs [J]. Water Air & Soil Pollution Focus, 2001,1(5/6):303-312.
[16] Collett J L, Bator A, Sherman D E, et al. The chemical composition of fogs and intercepted clouds in the United States [J]. Atmospheric Research, 2002, 64(1-4):29-40.
[17] Haeffelin M, Bergot T, Elias T, et al. Parisfog: shedding new light on fog physical processes [J]. Bulletin of the American Meteorological Society, 2010,91(6):767-783.
[18] Stolaki S, Haeffelin M, Lac C, et al. Influence of aerosols on the life cycle of a radiation fog event. A numerical and observational study [J]. Atmospheric Research, 2015,151:146-161.
[19] 李子华,彭中贵.重庆市冬季雾的物理化学特性[J]. 气象学报, 1994, 52(4):477-483.
Li Z H, Peng Z G. Physical and chemical characteristics of the Chongqing winter water fog [J]. Acta Meteorological Sinica, 1994, 52(4):477-483.
[20] 李子华,董韶宁,彭中贵.重庆雾水化学组分的时空变化特征[J]. 南京气象学院学报, 1996,19(1):63-68.
Li Z H, Dong S N, Peng Z G. Spatial/temporal variabilities of chemical constituents fog water sampled in Chongqing [J]. Journal of Nanjing Institute of Meteorology, 1996,19(1):63-68.
[21] 李 一,张国正,濮梅娟,等.2006年南京冬季浓雾雾水的化学组分 [J]. 中国环境科学, 2008,28(5):395-400.
Li Y, Zhang G Z, Pu M J, et al. The chemical composition of fog water in the winter of 2006 of Nanjing [J]. China Environmental Science, 2008,28(5):395-400.
[22] 刘红杰,王 玮,高金和,等.闽南地区酸性雾水特征初探[J]. 环境科学研究, 1996,9(5):30-32.
Liu H J, Wang W, Gao J H, et al. Preliminary study on the characteristics of acid fog in Minnan area [J]. Research of Environment Sciences, 1996,9(5):30-32.
[23] 吴 兑.南岭大瑶山浓雾雾水的化学成分研究[J]. 气象学报, 2004, 62(4):476-485.
Wu D. The study on fog-water chemical composition in Dayaoshan of Nanling Mountain [J]. Acta Meteorological Sinica, 2004,62(4):476- 485.
[24] 顾凯华,樊曙先,黄红丽,等.南京冬季雾天颗粒物中PAHs分布与气象条件的关系 [J]. 中国环境科学, 2011,31(8):1233-1240.
Gu K H, Fan S X, Huang H L, et al. Characteristics of polycyclic aromatic hydrocarbons (PAHs) in particles and the influence of foggy weather conditions during the winter in Nanjing [J]. China Environmental Science, 2011,31(8):1233-1240.
[25] 徐 峰,牛生杰,张 羽,等.湛江东海岛春季海雾雾水化学特性分析 [J]. 中国环境科学, 2011,31(3):353-360.
Xu F, Niu S J, Zhang Y, et al. Analyses on chemical characteristic of spring sea fog water on Donghai island in Zhanjiang, China [J]. China Environmental Science, 2011,31(3):353-360.
[26] 文 彬,银 燕,秦彦硕,等.2009年夏季黄山云雾水化学特征及来源分析 [J]. 中国环境科学, 2012,32(12):2113-2122.
Wen B, Yin Y, Qin Y S, et al. Analyses of chemical characteristic in fog/cloud water and source at Mount Huangshan in summer 2009 [J]. China Environmental Science, 2012,32(12):2113-2122.
[27] Twomey S. Pollution and the planetary albedo [J]. Atmospheric Environment, 1974,8:1251-1256.
[28] Twomey S. The influence of pollution on the shortwave albedo of clouds [J]. Journal of The Atmospheric Sciences, 1977,34:1149-1152.
[29] Izhar S, Gupta T, Panday A K. Scavenging efficiency of water soluble inorganic and organic aerosols by fog droplets in the Indo Gangetic Plain [J]. Atmospheric Research, 2019:1-41.
[30] Wang Y, Guo J, Wang T, et al. Influence of regional pollution and sandstorms on the chemical composition of cloud/fog at the summit of Mt. Tai shan in northern China [J]. Atmospheric Research, 2011,99: 434-442.
[31] 孙 玉,樊曙先,张 健,等.南京2013年冬季三级分粒径雾水化学特征 [J]. 中国环境科学, 2015,35(4):1019-1031.
Sun Y, Fan S X, Zhang J, et al. Chemical characteristics of the three- stage fog water in the winter of 2013 in Nanjing [J]. China Environmental Science, 2015,35(4):1019-1031.
[32] 朱丹丹,樊曙先,胡春阳,等.南京三级分档雾水有机酸和无机组分化学特征 [J]. 中国环境科学, 2020,40(8):3342-3351.
Zhu D D, Fan S X, Hu C Y, et al. Chemical characteristics of organic acids and inorganic components in Nanjing three-stage fog water [J]. China Environmental Science, 2020,40(8):3342-3351.
[33] 康博识,樊曙先,张 悦,等.南京冬季持续性强浓雾天气中三级分档雾水的理化特性分析 [J]. 气象学报, 2017,75(2):356-370.
Kang B S, Fan S X, Zhang Y, et al. Physical and chemical characteristics of three-stage fog water in deep dense fog during the winter in Nanjing [J]. Acta Meteorologica Sinica, 2017,75(2):356- 370.
[34] Zhang S R, Fan S X, Wang Y, et al. Chemical characteristics of size-resolved fog water at an urban site in Nanjing and the summit of Mt. Lu, East China [J]. Atmospheric Environment, 2021,263:1-14.
[35] 银 燕,童尧青,魏玉香,等.南京市大气细颗粒物化学成分分析 [J]. 大气科学学报, 2009,32(6):723-733.
Yin Y, Tong Y Q, Wei Y X, et al. Analysis of chemical composition of atmospheric fine particles in Nanjing [J]. Transactions of Atmospheric Sciences, 2009,32(6):723-733.
[36] 王红磊,朱 彬,马梁臣,等.南京市夏季城市不同功能区气溶胶污染特征 [J]. 南京信息工程大学学报(自然科学版), 2010,2(3):221-225.
Wang H L, Zhu B, Ma L C, et al. Characteristics of aerosol pollution in different functional areas of Nanjing in summer [J]. Journal of Nanjing University of Information Science & Technology, 2010,2(3): 221-225.
[37] 张秋晨,朱 彬,苏举峰,等.南京3类不同大气污染过程下气溶胶水溶性无机离子的特征研究 [J]. 环境科学, 2012,33:1945-1951.
Zhang Q C, Zhu B, Su J F, et al. Study on the characteristics of aerosol water-soluble inorganic ions under three different air pollution processes in Nanjing [J]. Environmental Science, 2012,33: 1945-1951.
[38] Wang H L,Zhu B, Shen L J, et al. Size distributions of aerosol and water-soluble ions in Nanjing during a crop residual burning event [J]. Journal of Environmental Sciences, 2012,24:1457-1465.
[39] 李晓娜,黄 健,申双和,等.一次高压型海雾中的液态含水量演变特征 [J]. 热带气象学报, 2010,26(2):79-85.
Li X N, Huang J, Shen S H, et al. Evolution characteristics of liquid water content in a high pressure sea fog [J]. Journal of Tropical Meteorology, 2010,26(2):79-85.
[40] 苏 捷,赵普生,陈一娜.北京地区不同天气条件下气溶胶数浓度粒径分布特征研究[J]. 环境科学, 2016,37(4):1208-1218.
Su J, Zhao Pu S, Chen Y N. Study on aerosol number concentration and particle size distribution under different weather conditions in Beijing [J]. Environmental Science, 2016,37(4):1208-1218.
[41] Collett Jr J L, Hoag K J, Sherman D E, et al. Spatial and temporal variations in San Joaquin Valley fog chemistry [J]. Atmospheric Environment, 1999,33:129-140.
[42] Collett Jr J L, Iovinelli R, Demoz B. A three-stage cloud impactor for size-resolved measurement of cloud drop chemistry [J]. Atmospheric Environment, 1995,29:1145-1154.
[43] 中国气象局.地面气象观测规范 [M]. 北京:气象出版社, 2003:32- 34.
China Meterological Administration. Code for surface meteorological observation [M] Beijing: Meteorological Press, 2003:32-34.
[44] 王 元,牛生杰,陆春松,等.西双版纳热带雨林地区冬季辐射雾理化特征的观测研究 [J]. 中国科学:地球科学, 2021,51:1-14.
Wang Y, Niu S J, Lu C S, et al. Observational study of the physical and chemical characteristics of the winter radiation fog in the tropical rainforest in Xishuangbanna, China [J]. Science China Earth Sciences, 2021,51:1-14.
[45] Niu S J, Liu D Y, Zhao L J, et al. Summary of a 4-year fog field study in northern Nanjing, part 2: Fog microphysics [J]. Pure and Applied Geophysics, 2012,169:1137-1155.
[46] 郭丽君,郭学良,方春刚,等.华北一次持续性重度雾霾天气的产生、演变与转化特征观测分析 [J]. 中国科学: 地球科学, 2015,45:427- 443.
Guo L J, Guo X L, Fang C G, et al. Observation analysis on characteristics of formation, evolution and transition of a long-lasting severe fog and haze episode in North China [J]. Science China: Earth Sciences, 2015,58:329-344.
[47] Hussein T, Hämeri K, Aalto P, et al. Particle size characterization and the indoor-to-outdoor relationship of atmospheric aerosols in Helsinki [J]. Scandinavian Journal of Work Environment & Health, 2004, 30(suppl 2):54-62.
[48] 樊曙先,黄红丽,顾凯华,等.雾过程对大气气溶胶PM10中多环芳烃粒径分布的影响[J]. 高等学校化学学报, 2010,31(12):2375-2382.
Fan S X, Huang H L, Gu K H, et al. Effect of fog process on particle size distribution of polycyclic aromatic hydrocarbons in atmospheric aerosol PM10[J]. Chemical Journal of Chinese Universities, 2010, 31(12):2375-2382.
[49] 王 庆,樊明月,李 季,等.济南冬季雾的微物理结构及其对能见度的影响 [J]. 大气科学, 2021,45(2):333−354.
Wang Q, Fan M Y, Li J, et al. The microphysical characteristics of winter fog in Jinan and its effect on visibility [J]. Chinese Journal of Atmospheric Sciences, 2021,45(2):333−354.
[50] 唐孝炎,张远航,邵 敏.大气环境化学 [M]. 北京:高等教育出版社, 2006:268-327.
Tang X Y, Zhang Y H, Shao M. Atmospheric environmental chemistry [M]. Beijing: Higher Education Press, 2006:268-327.
[51] Petters M D, Kreidenweis S M. A single parameter representation of hygroscopic growth and cloud condensation nuclei activity [J]. Atmospheric Chemistry And Physics, 2007:1961-1971.
[52] Moore K F, Sherman D E, Reilly J E, et al. Drop size-dependent chemical composition in clouds and fogs [J]. Part Ⅰ Observations. Atmospheric Environment, 2004,38:1389-1402.
[53] Zhang F, Li Z, Li Y, et al. Impacts of organic aerosols and its oxidation level on CCN activity from measurement at a suburban site in China [J]. Atmospheric Chemistry And Physics, 2016,16:5413-5425.
[54] Kuang Y, He Y, Xu W Y, et al. Distinct diurnal variation in organic aerosol hygroscopicity and its relationship with oxygenated organic aerosol [J]. Atmospheric Chemistry And Physics, 2020,20:865-880.
[55] Gilardoni S, Massoli P, Giulianelli L, et al. Fog scavenging of organic and inorganic aerosol in the Po Valley [J]. Atmospheric Chemistry And Physics, 2014, 14:6967-6981.
[56] Lu C S, Niu S J, Tang L L, et al. Chemical composition of fog water in Nanjing area of China and its related fog microphysics [J]. Atmospheric Research, 2010,97:47-69.
[57] Chakraborty A, Bhattu D, Gupta T, et al. Real-time measurements of ambient aerosols in a polluted Indian city: Sources, characteristics, and processing of organic aerosols during foggy and nonfoggy periods [J]. Journal of Geophysical Research-Atmospheres, 2015,120:9006-9019.
[58] Zhang Q, Jimenez J L, Worsnop D R, et al. A case study of urban particle acidity and its influence on secondary organic aerosol [J]. Environmental Science & Technology, 2007,41:3213-3219.
[59] Sasakawa M, Uematsu M. Chemical composition of aerosol, sea fog, and rainwater in the marine boundary layer of the northwestern North Pacific and its marginal seas [J]. Journal of Geophysical Research-Atmospheres, 2002,107:1-179.
Effects of Nanjing fog process on the spectral distribution and chemical composition of atmospheric aerosols.
ZHANG Si-rui1,2,3, FAN Shu-xian1,2*, WANG Yuan1,4, HU Chun-yang1,5, ZHANG Hong-wei1, ZHU Dan-dan1, GE Pan-yan1,2
(1.Collaborative Innovation Center on Forecast and Evaluation of Meteorological Disasters, Key Laboratory for Aerosol-Cloud-Precipitation of China Meteorological Administration, Nanjing University of Information Science & Technology, Nanjing 210044, China;2.School of Atmospheric Physics, Nanjing University of Information Science and Technology, Nanjing 210044, China;3.Shangrao Meteorological Bureau, Shangrao 334000, China;4.Collaborative Innovation Center for Western Ecological Safety, Lanzhou University, Lanzhou 730000, China;5.Unit 94582 of People's Liberation Army of China, Queshan 463217, China)., 2022,42(11):4961~4973
In order to explore the effects of fog process on the chemical composition and size distribution of aerosol particles in Nanjing. Three-stage fog samples and particle-size aerosol samples were collected at the same time in the fog observation in winter 2017. The microphysical quantity and aerosol spectral distribution of fog, the chemical composition of three-stage fog samples and particle-size aerosol samples before, in and after fog were compared and analyzed. The results showed that: in the winter of 2017, the fog droplet liquid water content of the first fog process in Nanjing was in the shape of asymmetric "V" with the particle size distribution, and the lowest value was located at 7μm. The fog droplet liquid water content in the second fog process was a 3peak shape with the particle size distribution, and the peaks were located at 5, 15and 21.5μm. The mass concentration of small particle size aerosol decreased while that of large particle size aerosol increased in the stage of fog formation and development. The mass concentration of aerosol particles reached the lowest in the whole particle size in the stage of fog maturity, and the mass concentration of larger particle size aerosol decreased significantly. Compared with that before fog, the peak value of aerosol mass concentration after fog moved to the direction of large particle size. Before fog, the water-soluble ion components in aerosol were enriched in small particles with particle size < 0.43μm, with the progress of fog process, nucleation and hygroscopic growth lead to the enrichment of water-soluble ions to larger particle size. The newly generated aerosol in the fog was released with the evaporation of fog droplets, resulted in the increase of NO3-, SO42-and NH4+concentrations after fog. The neutralization rate of aerosol with smaller size was higher, and the new droplets in the early stage of fog formation were more acidic. With the gradual neutralization of fog process, the pH value of fog water increased.
fog microphysical structure;chemical composition;fog and aerosol;particle size distribution
X513
A
1000-6923(2022)11-4961-13
张思蕊(1991-),女,陕西渭南人,助理工程师,硕士,主要从事大气化学研究.发表论文3篇.
2022-04-15
国家自然科学基金资助项目(42075066,41675132,42075063);国家重点研发计划项目(2018YFC1507905);江苏省研究生科研创新计划项目(KYCX21_0970);国家自然科学基金青年科学基金项目(42205072)
* 责任作者, 教授, shuxianf@nuist.edu.cn
猜你喜欢
成都信息工程大学学报(2022年3期)2022-07-21
中国土壤与肥料(2021年5期)2021-12-02
成都信息工程大学学报(2021年2期)2021-07-22
成都信息工程大学学报(2021年2期)2021-07-22
辐射防护通讯(2019年3期)2019-04-26
军事文摘·科学少年(2018年4期)2018-09-29
奥秘(创新大赛)(2018年6期)2018-08-24
中成药(2016年4期)2016-05-17
中国光学(2015年5期)2015-12-09
国外科技新书评介(2014年4期)2014-12-17
