
取代 - 2 - 苯基异噻唑啉酮化合物的定量构效关系研究
2022-12-07 07:40:04吴来明蔡兰坤
材料保护 2022年9期
杨 星,闫 莹,2,周 浩,吴来明,蔡兰坤
(1. 华东理工大学资源与环境工程学院 国家环境保护化工过程环境风险评价与控制重点实验室,上海 200237;2. 中国科学院海洋研究所 山东省腐蚀科学重点实验室,山东 青岛 266000;3. 上海博物馆 馆藏文物保存环境国家文物局重点科研基地,上海 200050)
0 前 言
异噻唑啉酮类化合物是一类具有广谱杀菌性能的非氧化性杀菌剂[1],其具有抑菌能力强、应用剂量小、配伍性好、低毒、在环境中不富集、能够快速降解等优点,对细菌、真菌、虫类以及海藻类均具有较强的杀活性[2]。早在20世纪80年代,异噻唑啉酮类化合物就广泛用于海洋防污、工业水处理、医疗、胶黏剂、建筑材料等领域[3-6]。为得到更绿色,杀菌活性更高的异噻唑啉酮类化合物,国内外研究学者对其结构进行不断地改造。Khalaj等[2]合成并测试了21种取代苯基异噻唑啉酮化合物对6种微生物的抗菌活性,发现当苯环上存在吸电子基时对革兰氏阳性菌的杀菌效果优于革兰氏阴性菌。王向辉等[7]采用“一锅法”合成了8种2 - (苯并异噻唑啉 - 3 - 酮 - 2 - 基)甲酸酯类化合物,发现该类化合物药物浓度为100 mg/L时对枯燥芽孢杆菌和金黄色葡萄球菌的抑菌效果为100%。张佩玉等[8]合成了10个2 - 取代异噻唑啉酮化合物,实验发现在噻唑环上引入较多的卤素原子,其生物活性有明显的提高。
近年来,越来越多的研究者利用定量构效关系研究(QSAR)方法来研究新型化合物的分子结构与生物活性之间的关系[9-13]。定量构效关系可以利用理论计算和统计分析工具来研究化合物结构(包括二维结构、三维结构和电子结构)与其生物活性、遗传毒性等之间的定量关系[14,15],已成为解释和预测化学化合物性质和活性以及物质合成的重要工具[16]。作为环境友好型抗菌剂,近年来对异噻唑啉酮类化合物的合成与杀菌性能研究的报道居多。而针对研究异噻唑啉酮类化合物的分子结构与杀菌活性之间的关系的研究较少。Morley等[17,18]通过使用半经验和从头算分子轨道方法评估一系列结构多样的3 - 异噻唑酮抑制大肠杆菌生长所需的最小抑菌浓度,研究发现3 - 异噻唑酮衍生物的实验活性与分子的几何形状、电子性质或前沿轨道能之间没有显著的相关性。冯文等[19]采用半经验算法对11个Mannich碱类苯丙异噻唑啉酮化合物进行构效关系研究,研究结果表明当取代基为供电子基团时,MIC50和ELUMO、EL-H、LS-N、QN以及二面角D之间呈现良好的相关性。
为了进一步研究异噻唑啉酮类化合物结构与生物活性之间的构效关系,本工作选取了10种不同基团取代的苯基异噻唑啉酮化合物为研究对象,采用量子化学密度泛函理论B3LYP计算方法中6-311G(d,p)基组对分子结构进行优化[20],计算得到化合物的几何结构、电子结构、分子性质等量子化学参数,通过多元回归分析,筛选影响这类化合物抗菌活性的主要因素,并建立相应的定量构效关系模型,所得模型对预测、设计和研究新型高效、低毒环保的异噻唑啉酮杀菌剂有较高的参考价值。
1 研究方法
1.1 活性参数的建立
本研究以取代 - 2 - 苯基异噻唑啉酮类化合物为研究对象,从文献[21]中获取了一系列10种取代 - 2 - 苯基异噻唑啉酮衍生物(见图1)的分子结构及其对大肠杆菌的抑菌活性浓度。其中,活性数据以对大肠杆菌的最低抑制浓度(MIC50)表示,在QSAR研究中将MIC50的单位标准化为mol/L,以log(1/MIC)表示化合物的抗菌活性。这10种化合物标准化后的活性参数log(1/MIC)值列于表1中,供后续建立定量构效关系的模型使用。
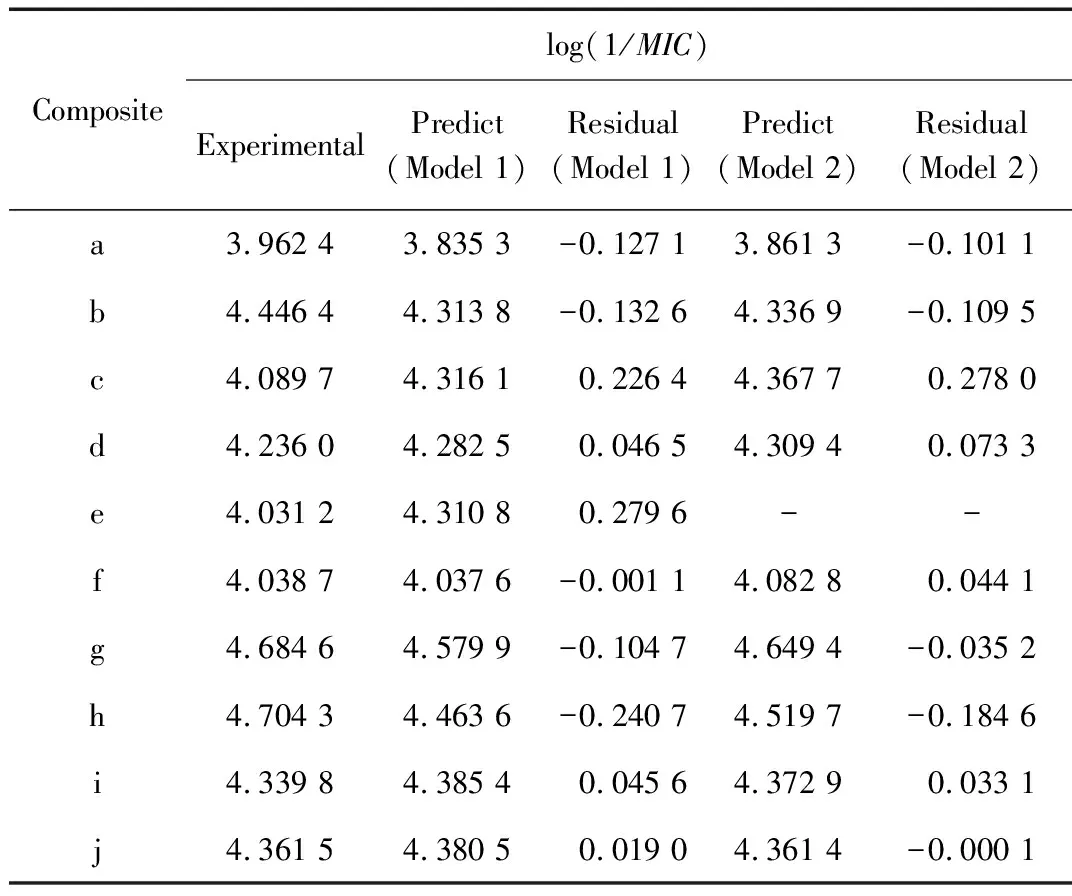
表1 取代 - 2 - 苯基 - 异噻唑啉酮化合物抗菌活性的实验值和预测值
1.2 化合物结构参数获取
本研究采用Gaussview软件构建化合物的分子模型,采用密度泛函理论(DFT)的Becke三参数混合泛函方法(B3LYP)对10种取代 - 2 - 苯基异噻唑啉酮化合物分子结构进行优化,在构型优化基础上进行了振动频率计算,确认所得构型为稳定构型,以获得本研究中所有化合物的最低能量[11],然后计算得到量子化学参数和物理化学参数,所有计算通过Gaussian09量子化学程序包进行。
1.3 参数的获取与筛选
为确保所建定量构效关系模型的科学性、可靠性,本研究从量子化学计算结果当中选取了以下几个参数: S-N键级(BS-N)、最高占据轨道能量(EHOMO)、最低空轨道能量(ELUMO)、LUMO和HOMO的能量差(EL-H)、异噻唑啉酮主环上S和N的原子净电荷Q(S)和Q(N),分子的偶极矩(μ)、异噻唑啉酮环和苯环之间的二面角(D)以及化合物的辛醇 - 水分配系数(lgP)。利用统计学原理,采用SPSS Statistics 26对目标化合物活性参数与分子结构参数进行相关性分析,同时考察了各个结构参数与化合物抗菌活性之间的关系以及各个参数之间的共线性关系,筛选出影响目标化合物抑菌活性的主要结构参数。
1.4 QSAR模型建立和回归分析
以上述筛选的影响目标化合物抑菌活性的主要结构参数为自变量,抑菌活性参数log(1/MIC)为因变量,采用数学分析软件SPSS Statistics26进行多元回归分析,通过各影响因素的相关系数,确定该类化合物的定量构效关系模型,根据抑菌活性参数获得对应化合物的预测值(表1)。
2 结果与讨论
2.1 取代 - 2 - 苯基 - 异噻唑啉酮化合物的分子结构参数
对所选10种2 - 苯基异噻唑啉酮类化合物经过结构优化后,所得到的相关分子结构量子化学参数如表2所示。根据计算所得的量子化学参数可知,异噻唑啉酮环中S-N键的键级最小,因此异噻唑啉酮环上S-N键的稳定性最差、最容易被进攻从而断裂。结合异噻唑啉酮类化合物的杀菌机理可知,S-N键越弱,便越容易断裂,越容易与微生物中的含S物质发生反应,形成S-S键,进而灭菌效果更出色,异噻唑啉酮类化合物的杀菌性能更好。
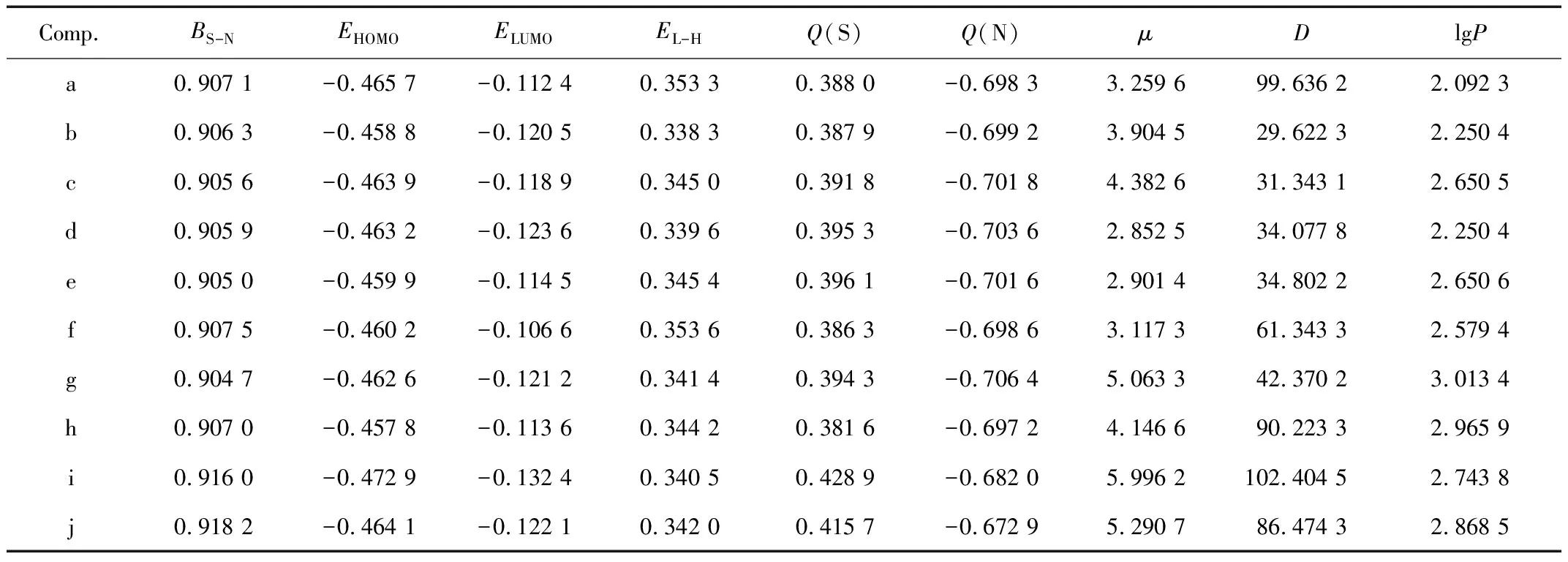
表2 异噻唑啉酮化合物的量子化学参数
由异噻唑啉酮主环上S原子和N原子净电荷的数值分布可以看出,不同的官能团会对不同化合物的原子净电荷产生一定的影响。在本研究中,i组和j组异噻唑啉酮化合物的S(1)电位所携带正电荷最多,N(2)电位携带负电荷最少,但是它们的抗菌性能在所选的10组异噻唑啉酮类化合物中并不突出。可能存在的原因是取代基团的影响:i组取代基团中,异噻唑啉酮环和苯环之间由一个亚甲基相连,因此后面苯环中C的位置也和其他几组化合物的不同,推测其中的原因可能为:亚甲基取代基团致使i组异噻唑啉酮化合物的杀菌性能下降,因为亚甲基的存在,异噻唑啉酮环上非氢原子的电荷数量减少,进而影响了化合物的活性。j组化合物中苯环对位取代基团为乙酰基,乙酰基是吸电子基团(g组主要是由于其取代基的电负性较大,使得苯环周边电子密度降低,本工作中电负性不作为主要考虑因素),当乙酰基取代苯环上的氢后,苯环上的电子云密度会随之降低,j组的异噻唑啉酮化合物也因此受到影响,从而导致其杀菌性能下降。
2.2 活性参数与结构参数的相关性分析
对活性参数与量子化学参数进行相关性分析,为确保所建模型的稳定性,考察了各个结构参数与化合物抗菌活性之间的关系以及各个参数之间的共线性问题,表3为由相关分析得到的活性参数与各结构参数之间的相关系数(一般来说,0~0.090为不相关,0.100~0.300为弱相关,0.300~0.500为中等相关,0.500~1.000为强相关。)
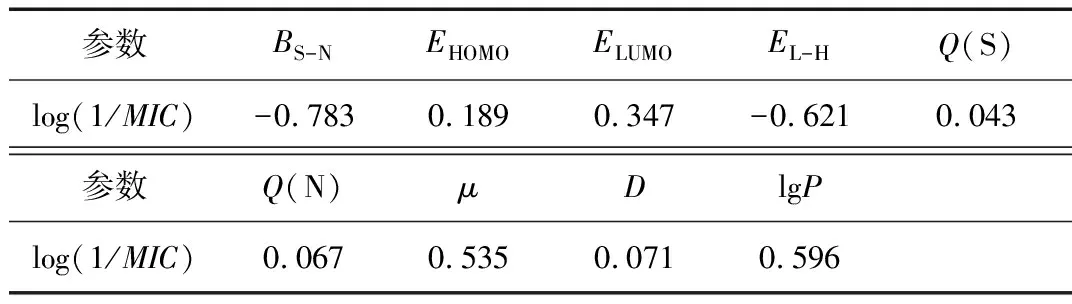
表3 取代化合物的活性参数与结构参数相关性分析
由表3可以看出,异噻唑啉酮主环上S-N键的键级(BS-N)、最低空轨道与最高占据轨道的能量差(EL-H)以及化合物的辛醇-水分配系数(lgP)是影响取代 - 2 - 苯基异噻唑啉酮化合物抑制大肠杆菌活性的主要因素,从相关性可以看出, lgP值越大,EL-H、BS-N值越小,化合物的抑菌效果越好,这是因为S-N键是异噻唑啉酮环中最不稳定的键,当化合物穿过受体的细胞壁和细胞膜后,与细胞内含S的蛋白质、酶等物质相互作用,形成更为稳定的二硫键(S-S),从而破坏受体活性,抑制受体的生长[22,23]。而物质的lgP以及分子的EL-H是亲脂性或疏水性的重要指标,lgP值越大,EL-H越小,表示物质的脂溶性越大,导致物质进入生物膜中不易流通,降低生物的活性[24]。此外,从表中可看出,不同取代化合物S原子、N原子的净电荷和二面角的改变几乎不影响其抑菌活性。
2.3 QSAR模型的建立
根据选取10种取代 - 2 - 苯基异噻唑啉酮的样本活性参数log(1/MIC)为因变量,以及由相关分析筛选的受主要影响的3种结构参数BS-N、EL-H、lgP为自变量,进行多元回归分析(MLR),得到模型(1)如下:

(1)
式中,n为样本数,n=10;R为复相关系数,R=0.794;R2为判定系数,R2=0.630;Se为样品标准残差,Se=0.243;F为方差检验值,F=3.408;sig.为显著性,sig.=0.094。通常情况下,R,R2,F越大,Se越小,代表模型拟合越好,更能够准确地预测该类化合物的活性[25]。将取代 - 2 - 苯基异噻唑啉酮化合物的3种主要影响因素代入到模型(1)中,可得到由模型(1)预测的活性参数log(1/MIC)值及实验值与预测值的残差(表1),与模型(1)的标准残差相比,化合物e的残差高于模型(1)的标准残差,故将化合物e作为奇异点舍掉。将其余9种取代 - 2 - 苯基异噻唑啉酮重新进行QSAR建模,得到模型(2)如下:
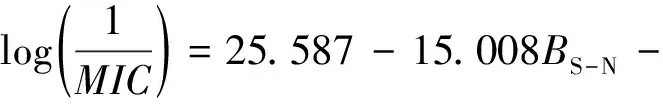
(2)
n=9,R=0.864,R2=0.747,Se=0.169,F=4.917,sig.=0.059。
模型(2)为舍弃奇异点后进行的线性回归分析,相比模型(1),模型(2)的R值,R2值以及F值均高于模型(1),且相比之下,模型(2)的标准残差值更小,这说明模型(2)更具备描述抗菌活性与分子结构的关系和预测新化合物的能力。同样将取代 - 2 - 苯基异噻唑啉酮化合物的3种主要影响因素代入到模型(2)中,可得到由模型(2)预测的活性参数log(1/MIC)值及实验值与预测值的残差(表1)。图2为取代 - 2 - 苯基异噻唑啉酮化合物抑制大肠杆菌活性的实验值与预测值散点图。由图2可得,模型(2)拟合良好,其相关系数高于模型1,具备较好的可预测性。
3 结 论
采用密度泛函理论和多元线性回归分析对10种取代 - 2 - 苯基异噻唑啉酮化合物的抑菌活性进行研究,筛选了影响该化合物的主要分子结构因素,建立了定量构效关系模型,结果表明:
(1)S-N键的键级(BS-N)、最低空轨道与最高占据轨道的能量差(EL-H)以及辛醇-水分配系数(lgP),是影响取代 - 2 - 苯基异噻唑啉酮化合物的抑菌活性的主要分子结构影响因素,减小分子的BS-N、EL-H,提高lgP均有利于增强取代 - 2 - 苯基异噻唑啉酮化合物的抑菌活性。
(2)建立的QSAR模型2具有良好的预测能力,可为新型异噻唑啉酮化合物的设计合成提供参考。
猜你喜欢
中学化学(2024年5期)2024-07-08 09:24:57
云南化工(2021年7期)2021-12-21 07:27:22
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:44
天然产物研究与开发(2018年7期)2018-08-21 02:04:02
天然产物研究与开发(2018年4期)2018-05-07 06:47:52
中学化学(2016年10期)2017-01-07 08:37:06
湖南城市学院学报(自然科学版)(2016年2期)2016-12-01 04:06:40
中国塑料(2015年10期)2015-10-14 01:13:13
郑州大学学报(工学版)(2015年1期)2015-03-24 00:55:36
天然产物研究与开发(2014年5期)2014-04-27 14:15:44
