
小团簇吸附活化原子氧的密度泛函研究
2022-12-03 08:55朱裔荣陈文昊颉雨佳
湖南工业大学学报 2022年5期
刘 叶,袁 佩,朱裔荣,陈文昊,颉雨佳
(1.湖南工业大学 材料与先进制造学院,湖南 株洲 412007;2.湘潭大学 化工学院,湖南 湘潭 411105)
1 研究背景
计算化学具有节约研究成本、缩短研究时间、预测新材料等特性,为开发新材料提供了理论依据。3d 过渡金属的物理和化学性质,决定了它在合金、锂电池、表面吸附和化学催化等方面的重要应用。黄钊文等[1]采用CASTEP(Cambridge Sequential Total Energy Package)程序,以Si-Co 合金作为锂离子电池负极材料,进行了分子设计模拟研究。结果表明,其导电性能会直接影响锂离子的输运和大倍率充放电性能,Si2Co具有较低的体积膨胀系数(71.151 5 %),以及较低嵌锂的形成能(0.457 1 eV),故具有较佳的电化学综合性能。Nong J.等[2]采用Dmol3程序,研究了Li-O2电池中氮掺杂碳基材料的结构与电子输运性能间的关系,发现未掺杂的单层石墨烯由于其电荷分布良好而没有活性位点吸附O2,因而吡啶-N 比石墨-N 对O2具有更高的催化活性。L.M.Jiménez-Díaz 等[3]采用Quantum Espresso 程序,对分子氧在Au12M(M 为 Cu、Ag、Ir)团簇上的吸附作用和解离作用进行了研究。其结果表明,金纳米团簇中添加过渡金属原子可以增强对CO 氧化反应的催化活性。Li T.T.等[4]采用VASP(Vienna Ab-initio Simulation Package)软 件,分 析 了12 种Cu13-mNim(m=0,1,13)团簇的结构,结果表明,对称性最高的Cu12Ni团簇比Cu13和Ni13团簇更易吸附O2和CO 分子,因而有利于CO 氧化。Lei X.等[5]采用Gaussian 09 软件,系统研究了分子氧和原子氧在Ag(100)表面氧化CO 的过程,结果表明,分子氧和原子氧对CO 氧化的活化能分别为28.8,8.78 kJ/mol,说明对CO 氧化都有很强的反应活性,但分子氧和原子氧在氧化过程中的氧化机理各不相同。Hao F.等[6]采用Dmol3程序,计算了环己基过氧化氢(cyclohexyl hydroperoxide,CHHP)的O—O 键长和变形能,结果表明钴负载的氮掺杂载体更容易使CHHP 解离,且计算结果与实验观测结果较吻合,实验得知催化剂钴(Co-N-rGO)催化环己烷的转化率为8.85%,选择性为85.73%。上述研究结果均表明,快速、环保的模拟计算可高效指导新材料的设计与合成[7]。
团簇可被作为介于原子与宏观物质之间的桥梁,是研究宏观催化体系的理想模型,不同尺寸的团簇可表现出不同于体材料的效果[8-9]。团簇的活性与结构、制备方法及载体等因素密切相关,识别团簇的真正活性位点变得非常困难。材料的研发成本和能耗较高、耗时较长等问题长期存在,为弥补材料合成中的问题,采用建模模拟计算材料的最优结构,指导改进合成现有的材料成分或设计新的催化剂显得尤为重要。设计高活性和高选择性的新型催化剂材料的关键,在于充分认识反应活性位点的结构与反应活性的关系。原子氧(O)和金属钴(Co)之间的相互作用是金属表面氧化、表面腐蚀及催化反应等过程中至关重要的影响因素,但已有文献中,对带电钴团簇吸附活化原子氧的研究较少。因此,本文拟采用密度泛函理论系统研究(n=1~5;q=0,+,-)团簇和团簇吸附原子氧后所得(n=1~5;q=0,+,-)团簇的几何结构、稳定性能和电子性质,考察电荷对原子氧吸附活化行为的影响,以期利用模拟计算结果为吸附、防腐蚀和催化应用(CO 氧化、烷烃氧化)等实验研究提供一定的理论指导。
2 计算方法
采用Dmol3程序包进行密度泛函理论计算,若无特殊说明,所有计算过程结果均由下列参数设置得到:自旋限制、广义梯度近似(general gradient approximation,GGA)的Perdew-Burke-Ernzerhof(PBE)泛函,其核处理方式采用全电子,基组为双数极化(double-numerial basis,DNP)。
几何优化中,所有结构都保持弛豫,对称性不限制,精度选择fine,位移和能量分别设为0.005 Å 和1×10-5Ha(1 Ha=27.211 6 eV),直至收敛。在此条件下计算O2键长,为1.227 Å,与实验值1.207 Å[7]间的误差较小。这一结果说明,所选用的方法和参数合理可靠。
通过文献分析[10-11],设计团簇结构,并进行优化,以获得具最低吸附能的结构。基于优化所得团簇不等价的可能顶位(T,top)、桥位(B,bridge)、空位(H,hole)位置上(见图 1),初始垂直距离大于3 Å 的位置处预置原子氧,再在相同条件下进行结构优化,并对所有优化后的结构(和)进行频率计算,无虚频[12],以确保结构是最低能量的稳定结构,之后计算其相应性质。
原子氧(O)吸附在团簇上的吸附能定义如下:
E(O)为原子氧的能量。
原子氧吸附在团簇上的吸附能Ea为负值,表示吸附放热,其绝对值越大,吸附能力越强。
3 结果与讨论
由图3 可以明显观察到,各个团簇的结合能均随着团簇尺寸的增加而增大,且随着n增大变化趋势逐渐趋于平缓,表明团簇的稳定性随团簇尺寸的增加逐渐增强。对比图3 中的曲线可以得知,阳离子型团簇()的平均结合能远远大于中性型()和阴离子型()团簇的结合能,这表明团簇失去一个电子后,可以显著增强该团簇的稳定性;而和团簇的结合能变化差异不大,但相较而言的结合能要稍低于的,这表明团簇得到一个电子后其稳定性略有降低。
最高占据分子轨道(highest occupied molecular orbital,HOMO)和最低未占据分子轨道(lowest unoccupied molecular orbital,LUMO)与化学反应活性有关。HOMO 是能量最高的轨道,可以提供电子以形成一个键,而LUMO 是能量最低的空轨道,该轨道上可以得到更多的电子。带隙能Egap为LUMO轨道与HOMO 轨道间的能量差值,是一个动力学稳定性指标,较大的带隙能意味着较高的动力学稳定性和较低的化学反应活性[8]。简而言之,带隙能可以反映电子被激发的难易程度以及被激发时所需能量的大小,其值越大,表示该分子越难以激发,分子活性越差。电荷对团簇最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)的能量影响顺序由大到小依次为负电荷、中性、正电荷。团簇的带隙能如图 4 所示。
由图8 所示各团簇的吸附能可以得知,除原子氧吸附后的Co2O T 团簇和Co2O B 团簇外,其余团簇的吸附能均随着原子数目的增加而呈现出规律性的变化,表现为,吸附能为负值表明吸附放热,其绝对值越大,吸附能力越强。由此说明,团簇对原子氧的吸附强度由大到小依次为。其中,所有结构中B 团簇对原子氧的吸附能力最强,其吸附能为-8.375 eV。对比中性团簇,负电荷的引入,有利于增强团簇对原子氧的吸附作用;而正电荷的引入,更容易削弱团簇对原子氧的吸附作用。
团簇的反应活性、稳定性,与电子结构和团簇中原子的数量和排列密切相关。因此,采用Mulliken电荷对原子之间的电荷转移进行分析。团簇中原子氧电荷的变化情况如图 10 所示,带隙能变化情况如图11 所示。
因原子氧的电负性(3.44)远大于钴原子的电负性(1.88),故原子氧对电子吸引的能力较强。钴团簇结构吸附原子氧之后,团簇中O 得电子而带负电,金属团簇失电子而带正电,电荷转移进一步表明团簇活化了原子氧[13]。
由图10 可看出,电荷对团簇吸附原子氧的作用呈现出规律性变化,金属团簇带负电时,O 原子与其作用最强,中性次之,金属团簇正电荷时作用最弱,氧原子电荷转移强度大小顺序为。相同的n数目条件下,带正电的团簇和中性团簇中,O 的得电子能力由小到大依次为顶位、桥位、空位;带负电的团簇,O 得电子的能力由大到小依次为顶位、桥位、空位。出现这一结果的原因,与团簇中相应吸附位置原子氧的配位数有关,O 原子与初始的团簇顶位、桥位和空位的配位数分别为1,2,3,经几何优化之后,部分结构(如Co2O T 团簇和Co3O B 团簇等)的配位数发生变化,与电荷协同作用的结果造成O 原子在团簇不同吸附位置处具有不同的得电子能力。
表1 O、和 B 的HOMO 和LUMO 轨道Table 1 HOMO and LUMO orbital of O, andO B
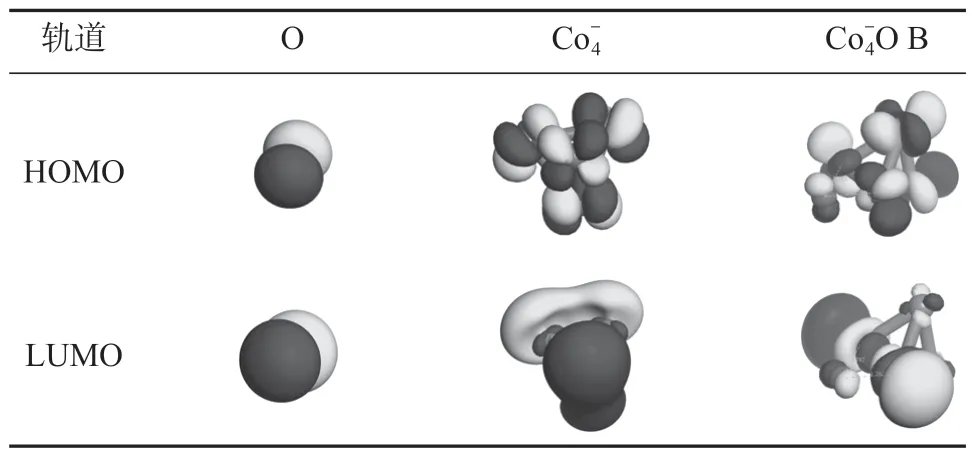
表1 O、和 B 的HOMO 和LUMO 轨道Table 1 HOMO and LUMO orbital of O, andO B
由表1 中的轨道情况可以看出,因O 是强电子接受体,其HOMO 轨道和LUMO 轨道都为p 型轨道;团簇为d 型轨道,其HOMO 轨道主要构成成键轨道,而LUMO 轨道主要由反键轨道构成。由B 团簇的HOMO 轨道和LUMO 轨道,可以明显观察到O 的2p 轨道与Co 的3d 轨道间的相互作用,这一结果进一步说明原子氧在团簇上的吸附主要是化学吸附。
4 结论
2)优化前的原子氧在初始位置相同的情况下,不同电荷的同种结构经过几何优化,原子氧会发生迁移并且被吸附到最稳定的吸附位点上。从吸附能的计算结果中可以看出,电荷的引入显著改变了团簇对原子氧的吸附强度。负电荷型团簇的吸附强度要比中性型团簇的强,而正电荷型团簇的吸附强度要比中性型团簇的弱,所有结构中,对原子氧的吸附能力最强的是带负电桥位型的B 团簇,其吸附能为-8.375 eV。
3)电荷对团簇吸附原子氧的作用呈现规律性变化,金属团簇带负电时,O 与其作用最强,中性团簇次之,正电荷团簇最弱。
5)当团簇带负电时,其Co—O 的键长要比中性团簇的长;而当团簇带正电荷时,其Co—O 的键长要比中性团簇的短。所有吸附结构中,吸附后的团簇中原子氧都获得电子而带负电,其中负电荷型团簇得电子最多,中性型团簇次之,正电荷型团簇最少。
本研究为原子氧活化阶段提供了相应的依据,结果有望为吸附、防腐蚀和催化应用(CO 氧化、烷烃氧化)等实验研究和工业生产提供理论指导,但涉及气敏、防腐蚀和氧化等体系,尚需进一步具体到理论研究和实验相互验证。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
皮肤病与性病(2021年3期)2021-07-30
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
中国外汇(2019年21期)2019-05-21
女友·家园(2017年7期)2017-07-19
新高考·高一物理(2016年7期)2017-01-23
米娜·女性大世界(2016年8期)2016-08-17
新高考·高一物理(2015年6期)2015-09-28
