
Single-cell RNA sequencing reveals the landscape of maize root tips and assists in identification of cell type-specific nitrate-response genes
2022-12-02 01:00XuhuiLiXingboZhngShuiGoFngqingCuiWeiweiChenLinFnYongwenQi
The Crop Journal 2022年6期
Xuhui Li,Xingbo Zhng,Shui Go,Fngqing Cui,b,Weiwei Chen,Lin Fn,Yongwen Qi,b,*
a Institute of Nanfan & Seed Industry,Guangdong Academy of Science,Guangzhou 510316,Guangdong,China
b College of Agriculture and Biology,Zhongkai University of Agriculture and Engineering,Guangzhou 510325,Guangdong,China
Keywords:Maize Single-cell RNA sequencing Root Nitrate
ABSTRACT The root system is fundamental for maize growth and yield.Characterizing its heterogeneity and cell type-specific response to nitrate at the single-cell level will shed light on root development and nutrient uptake.We profiled the transcriptomes of>7000 cells derived from root tips of maize seedlings grown on media with or without nitrate,and identified 11 major cell types or tissues and 85 cell type-specific nitrate-response genes,including several known nitrate metabolic genes.A pseudotime analysis showed a continuous pseudotime series with the beginning at meristematic zone cells and showed that the root hair cell was derived by differentiation of a subset of epidermal cells.Interspecies comparison of root cells between maize and rice revealed the conservation and divergence of the root cell types and identified 57,216,and 80 conserved orthologous genes in root hair,endodermis,and phloem cells respectively.This study provides a global view of maize root tip developmental processes and responses to nitrate at the single-cell level.The genes described in the present work could serve as targets for further genetic analyses and accurate regulation of gene expression and phenotypic variation in specific cell types or tissues.
1.Introduction
Maize plays a major role in global food,feed and fuel production,owing likely to its unique root system[1].The root system is fundamental for maize growth and yield.The primary root system is the first organ that emerges at germination,providing water and nutrients for the growing seedlings.In transverse section,primary roots are formed in two parts,the stele and the surrounding cortex.The stele comprises differentiated xylem vessels that function in water and nutrient transport and primary phloem elements that function in transport of photosynthate and other organic matter.The cortex consists of the endodermis,multiple layers of cortex tissue,exodermis,and epidermis connecting the root to the rhizosphere[2].The longitudinal structure includes the root cap at the terminal end and the apical meristematic zone(MZ),followed by the cell elongation zone in which newly formed cells elongate,and the differentiation zone[3].The epidermis of the differentiation zone is densely populated by tubular root hairs,which function in nutrient absorption[4].
Characterizing these differentiated tissues or cell types aims to elucidate their roles in shaping phenotypes.In previous studies[5-10],some tissues or root cell types of maize,including the stele,cortex,pericycle,and root hair,were dissected by hand or laser capture microdissection,and examined by transcriptome sequencing(RNA-seq).However,tissue dissection is labor-intensive and imprecise,given that it’s difficult to obtain enough target tissue and the tissue-or cell type-specific mRNA accumulation patterns tend to be masked by the incorporation of other cells.In recent years,single-cell RNA sequencing(scRNA-seq)has been developed and permits systematic capture and definition of cell features.scRNA-seq has been applied to samples from humans,mice,and plant tissues from Arabidopsis,rice,peanut,maize and other plants[11-26],revealing high heterogeneity in the given tissues and revealing the expression signatures of major cell types and developmental trajectories.These studies[27-29]showed the feasibility,challenges,and power of scRNA-seq in plants.
Increases in maize yield are linked largely to nitrogen(N)fertilizer use.However,N fertilizer capture by the roots of crop plants is relatively inefficient,with only 40%to 65%of applied N being used[30].Manipulation of root system development is an effective strategy for increasing yield and fertilizer use efficiency[9,31-33].Although the root system is essential to fertilizer uptake,economizing the costs for root development is pivotal as a resource-efficient strategy[34].Thus,an increased understanding of plant responses to N at the cell-type or tissue level will be crucial for the refinement of strategies to improve fertilizer uptake efficiency,agricultural productivity,and sustainability.The application of scRNA-seq to root development brings new hope for optimizing root nutrient uptake ability.Identification of root cell type-specific genes and their responses to nutrients,such as nitrate,might help researchers to fine-regulate root structure and absorption of nutrients under limiting conditions.
To characterize root tip cell heterogeneity at the single-cell level,in this study,we profiled the transcriptomes of single cells derived from maize root tips of seedlings grown on media with(nitrate+)or without nitrate(nitrate-).Most cell types were identified,and pseudotime analysis revealed some common initial cells for most of the cells and a continuous differentiation trajectory.To better understand and characterize the responses to nitrate in maize roots at single-cell resolution,we compared the single-cell transcriptome profiles of each cell type between the nitrate+and nitrate-data sets,and found that 85 genes were expressed and induced in a cell type-specific manner,four of which proved to be involved in nitrate absorption or utilization.Interspecies comparison of single-cell transcriptomes between maize and rice revealed the conservation and divergence of root cell types and their features.
2.Materials and methods
2.1.Plant material and growth conditions
Seedlings of the maize inbred line B73 were used for root scRNA-seq experiments.The dry seeds were sterilized with 1%sodium hypochlorite(NaClO)for five minutes,and washed three times with distilled water.Twenty sterilized seeds were placed on germination paper,which was soaked in nutrient solution(nitrate+)with 4 mmol L-1Ca(NO3)2,0.75 mmol L-1K2SO4,0.65 mmol L-1MgSO4,0.1 mmol L-1KCl,0.25 mmol L-1KH2PO4,10μmol L-1H3BO3,1μmol L-1MnSO4,0.1μmol L-1CuSO4,1μmol L-1ZnSO4,1 nmol L-1(NH4)6Mo7O24,and 0.1 mmol L-1FeNa-EDTA[35].The germination papers were then rolled and placed vertically in a sealed plastic bag.The paper roll was incubated at 25°C/22°C with an 8 h light/16 h dark cycle for 4 days.For nitrate deficiency treatment(nitrate-),the Ca(NO3)2in the nutrient solution was replaced by an equal concentration of CaCl2.
2.2.Protoplast isolation and single-cell RNA sequencing
Twenty apical sections(the bottom~2 cm)of primary roots were excised,cut into pieces of approximately 0.5 mm,and digested in protoplasting solution(1.25% cellulase R10,0.3%macerozyme R10,400 mmol L-1mannitol,20 mmol L-14-morpholineethanesulfonic acid,20 mmol L-1KCl,10 mmol L-1CaCl2,and 0.1%BSA).The mixture was placed in an incubator shaker at 75 r min-1for 80 min at room temperature,filtered twice through a 40-μm nylon strainer,and horizontally centrifuged at 70 g for 5 min.The supernatant was removed by pipette,and the remaining protoplasts were washed with washing solution(protoplasting solution without enzymes)[14,36].Protoplast viability and integrity were tested by trypan blue staining.The ratio of viable to total cells for each sample was>85%,and the concentration of protoplasts was adjusted to 1500 cellsμL-1with washing solution.
Approximately 10,000 root-tip protoplasts were loaded on a 10×Chromium Single Cell Instrument(10×Genomics,Pleasanton,CA,USA)to generate single-cell GEMs(gel bead-in-emulsion partition),followed by cell lysis and barcoded reverse transcription of the RNA in the droplets according to the user guide of the Chromium Single Cell 3′Reagent Kit v3.Quantification of the DNA library was performed with an Agilent 4200 Bioanalyzer(Agilent,Santa Clara,CA,USA),and the library was sequenced with an Illumina HiSeq2000 sequencer(Illumina,San Diego,CA,USA).
2.3.Bulked RNA-seq and pseudo-bulk analysis
To estimate the accuracy of scRNA-seq,bulked RNA-seq for primary root tips was performed.Total RNA was extracted from fourday-old root tips(2 cm)of primary roots from seedlings growing nitrate+and nitrate-using the RNAprep Pure Plant Kit(Qiagen),and each sampling was performed twice.RNA concentration and purity were measured with a NanoDrop 2000(Thermo Fisher Scientific,Wilmington,DE,USA).RNA integrity was assessed with the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system(Agilent Technologies,Palo Alto,CA,USA).A total of 1μg RNA per sample was used for sequencing libraries,which were generated with the NEBNext Ultra RNA Library Prep Kit for Illumina(NEB,Ipswich,MA,USA)following the manufacturer’s recommendations,and the libraries were sequenced on the Illumina NextSeq 500 sequencing platform.After removing the adapter and low quality reads,the remaining sequencing reads were aligned to the B73 reference genome with Hisat2 2.1.0[37].Gene expression values were counted with featureCounts v2.0.1[38]and differential expression analysis of count data was performed with the DESeq2 package[39](Padj≤0.05 and|log2fold change|≥1).To calculate the correlation between bulked RNA-seq and scRNA-seq counts,a pseudo-bulk count matrix was created by summing the read count values from each gene across all single cells.Read counts were then log2-transformed and Pearson’s correlation coefficients were calculated.
2.4.Data analysis of scRNA-seq of maize
To generate the gene expression matrix of single cells,the raw reads were aligned to the B73 reference genome,and gene expression was quantified using the Cell Ranger pipeline(version 3.1,10×Genomics).The B73 reference genome and annotation files were downloaded from Ensembl(ftp://ftp.ensemblgenomes.org/pub/plants/release-42).The gene annotation(gtf)file was filtered by the‘‘cellranger mkgtf”function with the‘‘--attribute=gene_bio type:protein_coding”argument.The‘‘cellranger mkref”function of Cell Ranger was used to build a reference.The‘‘cellranger count”function was performed to generate a raw count matrix.
2.4.1.Integrative analysis of two scRNA-seq datasets,cell clustering,and marker gene identification
Data preprocessing,dimensionality reduction,data integration,cell clustering,marker gene identification and visualization were performed mainly with the R package Seurat 4.0.1[40,41].The cells of two samples were filtered according to the criterion‘‘nFeature_RNA>400 & nFeature_RNA<5000 & percent.mt<5%& percent.cp<2.5%”,where percent.mt and percent.cp represent the respective percentages of mitochondrial and chloroplastexpressed genes.The two filtered count matrices were combined into one Seurat object with the function‘‘CreateSeuratObject”with‘‘min.cells=3,min.features=200”arguments.Then the‘‘rpca”(reciprocal PCA)algorithm was used to identify conserved anchors,and the two datasets were integrated with the‘‘FindIntegrationAnchors” and ‘IntegrateData” functions(dims=1:50).Further clustering and trajectory analyses were performed on the‘‘integrated”assay.The integrated dataset was scaled with the‘‘ScaleData”function.The 2000 most highly variable genes were used for principal component analysis(PCA)dimensionality reduction.Based on an elbow plot,the first 27 principal components(PCs)were used for cell clustering at a resolution of 0.5 according to a shared nearest neighbor(SNN)graph constructed with‘‘FindNeighbors”.The clusters were visualized and explored with nonlinear dimensionality reduction algorithms(UMAP)[42].Cluster-enriched genes(cluster-specific marker genes)were computed using the function‘‘FindAllMarkers”with options‘‘test.use=bimod”and‘‘logfc.threshold=1”.
To evaluate the impact of cell cycle in clustering,both filtered count matrices without cell-cycle-associated genes were subjected to integration,normalization,dimensionality reduction,and clustering with the same parameters as used above.The keywords(‘‘cell cycle”,‘‘cell division”,‘‘mitosis”,and‘‘mitotic”)were used to select cell cycle-associated genes according to their Gene Ontology(GO)function[43].
2.4.2.Cell-type annotation
To assign the cell clusters from scRNA-seq to known cell types,we used the following three strategies:1)expression of known cell type-specific marker genes collected from the literature(Table S1);2)calculated Pearson’s correlation coefficient between the transcriptome(2000 most highly variable genes)of each cell cluster and pericycle or root hair(bulked RNA sequencing)[5,10];and 3)RNA in situ hybridization(FISH).
To perform FISH,nine genes were selected according to marker genes identified by the‘‘FindAllMarkers”function.Probe synthesis was based on the mRNA sequence(Table S2)with FAM(6-carboxyfluorescein)and root tip tissues were harvested as described in section 2.1.The tissue samples were fixed with 4% paraformaldehyde above 12 h,followed by paraffin embedding and sectioning.After dewaxing and dehydration,the slices were digested with 20μg mL-1proteinase K at 37 °C for 20 min and hybridized with probes following Huang et al.[44].The cell nuclei were stained with DAPI(Servicebio,Wuhan,Hubei,China)for 8 min in the dark.DAPI glows blue at a UV excitation wavelength of 330-380 nm and an emission wavelength of 420 nm;FAM glows green at an excitation wavelength of 465-495 nm and an emission wavelength of 515-555 nm.Images were collected with a positive fluorescence microscope(Nikon Eclipse Ci,Tokyo,Japan).
2.4.3.Identification of cell type-specific nitrate-response genes
To identify cell type-specific nitrate-response genes,cells of each cell types were split into two putative clusters according to the treatments.Then‘‘FindMarkers”was used to find genes differentially expressed(DEGs)between two putative clusters on the assay of‘‘RNA”.A likelihood-ratio test(bimod)for single-cell gene expression was selected to assign differentially expressed genes at a cutoff of adjusted P-value<0.05 and|log2fold change|≥0.25.All differentially expressed genes from each cell type were assigned as cell type-specific nitrate-response genes.
2.4.4.Development trajectory and pseudotime analysis
For development trajectory analysis,the results of principal component analysis(PCA),nonlinear dimensionality reduction(UMAP),and cell clustering information obtained with the Seurat package was migrated to monocle3 0.2.3.0 with the‘‘as.cell_dat a_set”function of the SeuratWrappers 0.3.0 package with the‘‘assay=integrated”argument[45,46].To calculate the pseudotime of cells,a principal trajectory graph was fitted with the‘‘learn_-graph”function with default parameters for all cells except for root hair,epidermis,and root cap cells.MZ cells were designated as the root nodes of the trajectory graph and the pseudotime of each cell was calculated with the‘‘order_cells”function.The‘‘graph_test”function was used to identify trajectory-dependent genes at a cutoff of‘‘morans_I≥0.3 and q_value≤0.05”.
To characterize the developmental trajectory of the root hair and epidermis,pseudotime analysis and visualization was performed with dyno 0.1.2.First,the integrated expression data of root hair and epidermis in Seurat object was extracted with the‘‘wrap_expression”function.Second,developmental trajectory was inferred with the‘‘infer_trajectory”function by the method of‘‘slingshot”[47,48].Then two branches were identified.To identify genes with branch-specific expression,the gene importance for the whole trajectory was measured with the‘‘calculate_overall_fea ture_importance”function,and the top 100 genes were hierarchically clustered and used to generate a heat map with the‘‘plot_heatmap”function with‘‘features_oi=100”.
2.5.Interspecies analysis of root scRNA-seq data from maize and rice
Three rice root scRNA-seq read count matrices were collected for interspecies comparison[16,25].The relationships between orthologous genes between maize and rice were obtained from MaizeGDB(https://maizegdb.org/).A set of 7996 orthologous gene pairs were retained for dataset integration and further analysis(DataSet 1).In order to ensure the balance and applicability among five datasets(three rice datasets and two maize datasets),approximately 3500 rice root tip cells from three datasets(scRNA-seq data from 9311,Nipponbare and ZH11)were randomly sampled according to the initial proportion of a given cell type.The three subsets of data thus contained about 3500 cells with all the captured cell types and in proportions similar to those in each original dataset.The datasets of rice and maize were then integrated with the‘‘rpca”algorithm with dims=1:30,followed by‘‘ScaleData”and‘‘RunPCA”[40,41,49].Using an elbow plot,the top 24 PCs were selected for UMAP dimensionality reduction and shared nearest neighbor(SNN)graph construction.Clustering was performed on the integrated assay with resolution of 0.6.Mean expression values of each cell type were log2-transformed and Pearson’s correlations were calculated to compare maize and rice.
To identify conserved genes in root hair,endodermis,and phloem,root hair cells from nitrate+,nitrate-,9311,Nipponbare and ZH11(Seurat_cluster 14),endodermis cells from nitrate+,nitrate-,9311,and Nipponbare,and phloem cells from nitrate+,nitrate-and ZH11 were selected,and the corresponding gene expression matrices were generated.The 500 orthologous genes with highest variance across cells in each matrix were then hierarchically clustered using the ward.D2 method[50]and an orthologous gene cluster that displayed constitutive expression across almost all cells of the data set was selected as representing the genes conserved between maize and rice.
2.6.GO enrichment analysis
GO enrichment analyses and GO hierarchical graph construction were performed with AgriGO-v2[43].Significant GO categories were identified using a cutoff of FDR≤0.05.The GO enrichment results were visualized with the R package clusterProfiler[51].Significant GO categories were assigned according to the GO hierarchical graph and their shared genes.
3.Results
3.1.Single-cell RNA-seq of maize root tips and identification of cell types
To perform scRNA-seq on maize inbred line B73,sterilized seeds were germinated with nitrate(nitrate+)or without nitrate(nitrate-)for four days.The primary root length was greater under N-deficit than under N-sufficient conditions(Fig.S1).Approximately twenty apical root tips(bottom~2 cm)of primary roots from each growth condition were subjected to transcriptome profiling(Fig.1A).Transcriptomes were obtained for 3790 root cells with 156,184 reads and 1568 unique molecular identifiers(UMIs)per cell for nitrate+,and 3426 cells with 158,218 reads and 1635 UMIs per cell for nitrate-(Table S3).To assess the robustness of scRNA-seq and the effect of protoplasting,conventional bulked RNA-seq was performed on root tips without protoplasting,and strong correlations between the scRNA-seq psudo-bulked expression data and bulked RNA-seq expression data(Pearson’s correlation coefficient=0.8529 for nitrate+and 0.8500 for nitrate-)were observed(Fig.1B,C),a finding similar to that in Arabidopsis[14,21].Finally,two scRNA-seq expression matrices were integrated for cell type identification and further analysis(Fig.1D).
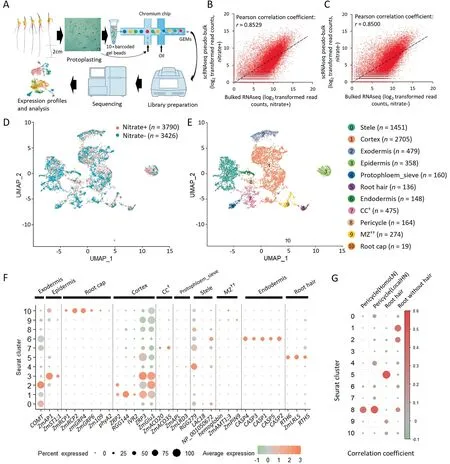
Fig.1.scRNAseq of maize root cells and identification of known cell types.(A)Workflow used for scRNAseq.(B,C)scRNAseq pseudo-bulked expression data were compared with bulk expression data of roots grown in nitrate+(B)and nitrate-medium(C).(D)UMAP dimensionality reduction plot of integrated scRNAseq data of nitrate+and nitrate,cells are colored by growth medium.(E)UMAP visualization of 11 cell cluster and cell types.(F)Expression pattern of known marker genes for each cell cluster.(G)Pearson’s correlation coefficient between pseudo-bulked expression data of each cluster and expression data of pericycle and root hair[5,10],and both dot diameter and color represent correlation coefficient.†,companion cells(CC)associated with the protophloem.††,meristematic zone.
To identify distinct cell populations of root tips,all the captured cells were classified into 11 major clusters with different cell numbers(Fig.1E;Table S4).To assign these clusters to known cell types,the following three strategies were used:1)The expression of known marker genes,which were,in general,highly specific to each cluster and known from previous studies,was observed(Table S1).For example,the epidermis marker genes ZmAP1 and ZmST1;1 were expressed specifically in cluster 3[52,53];The root hair marker genes,RTH6,ZmLRL5,and RTH5,were expressed preferentially in cluster 5[54-56].Smaller clusters were also readily annotated,with cluster 10 containing only 19 cells that specifically expressed ZmRCP1,ZmRCP2,ZmGRP4,ZmGRP6,Zm109 and phyA2,which were expressed specifically in root caps(Fig.1F)[57-60].The Casparian strip membrane domain proteins(CASPs)mark a membrane domain that predicts the formation of the Casparian strips in endodermis cells of Arabidopsis[61].The CASP orthologous genes of maize were expressed mainly in cluster 6(Fig.1F),suggesting that cells of this cluster were endodermal[17].Two homologs of marker genes of protophloem sieve cells in maize,ZmAPL and ZmLRD3[62,63],were expressed specifically in>10% of the cells of cluster 4.By this strategy,clusters 3-7,9,and 10 could be roughly assigned to the epidermis,protophloem sieve,root hair,endodermis,CC(companion cells associated with the protophloem),MZ,and root cap,respectively(Figs.1F,S2).2)Pearson’s correlation coefficients were calculated between single-cell expression data of each cluster and the bulked RNA-seq expression data of root cell types isolated by hand or laser-capture microdissection.Clusters 8 and 10 were assigned to pericycle and root hair cells(Fig.1G)[5,10].3)FISH was performed for clusters with few known marker genes or tissue-specific RNA-seq expression data.Thus,nine cluster-specific genes,identified by the FindMarkers function of Seurat,were selected for FISH(Table S2).Zm00001d049916 was significantly expressed in the stele(Fig.S3A),Zm00001d041611 in the epidermis(Fig.S3F),Zm00001d003418 in the endodermis (Fig.S3D),Zm00001d052842 in the exodermis near epidermis(Fig.S3B),Zm00001d032883 in the meristematic zone(MZ)(Fig.S3E),Zm00001d026163 in the cortex(Fig.S3G),Zm00001d051478 in pericycle cells (Fig.S3H).Zm00001d049606 and Zm00001d043344 were expressed mainly in the cell of the central cylinder near the phloem(Fig.S3C,I).In view of the expression of known marker genes,protophloem sieve and CC could be cautiously assigned to clusters 4 and 7,respectively.In summary,11 major cell types or tissues with sharply differing cell numbers were identified,representing almost all cell types and reflecting the high heterogeneity of maize root tips.
Because the cell cycle will influence cell gene expression patterns,affecting cell clustering[18],643 cell cycle-associated genes were removed from two datasets(DataSet 2).The cell clusters were similar to those of the cell cycle-associated genes,except for the endodermis(Fig.S4).Thus,the effect of cell cycle on clustering was limited.
3.2.Characteristic of marker genes of each cell type
Across the 11 cell types,871 cell type-specific marker genes were identified.These marker genes were defined as genes with more than a twofold expression difference between the tested cell types and the other cell types using‘‘FindMarkers”of Seurat(Data-Set 3).A large majority of cell type-specific marker genes were described as novel markers of specific cell types,as expected.But some known marker genes were also identified:RTH6 for root hair,CASP4 for endodermis,and ZmGRP4 for root cap(Figs.1F,S2;Data-Set 3).The top five marker genes showed highly specific expression in the corresponding cell types(Fig.S5).Three of the top five marker genes of the epidermis were simultaneously highly expressed in root hair.Two of the top five marker genes of the MZ were highly expressed in root cap.These findings were probably attributable to the similarity of the adjacent tissues.Two(Zm00001d051333 and Zm00001d008299)of the top five marker genes were highly expressed in the endodermis and exodermis.
To investigate the functional characteristics of each cell types or tissues,GO enrichment analysis for cell type-specific marker gene sets was performed and revealed the functions of cell groups,with the expression in almost all cell types or tissues except for the endodermis and pericycle being associated with stress response(Figs.2,S6;DataSet 4).In MZ cells,many genes involved in chromosomal duplication were identified.The marker genes of MZ were clearly enriched in the GO category for cell division processes.>50% of the marker genes for the pericycle encoded histone proteins.As the site of the initiation of lateral roots and two secondary meristems,pericycle cells usually display modest meristematic capacity.For root hair cells,their marker genes were,not unexpectedly,involved mainly in trichoblast and root hair cell differentiation.The marker genes of epidermis were associated mainly with ion transport and response to stimulus and stress.Marker genes of the cortex were involved in the jasmonic acidmediated signaling pathway,which mediates plant responses to biotic and abiotic stresses[64].
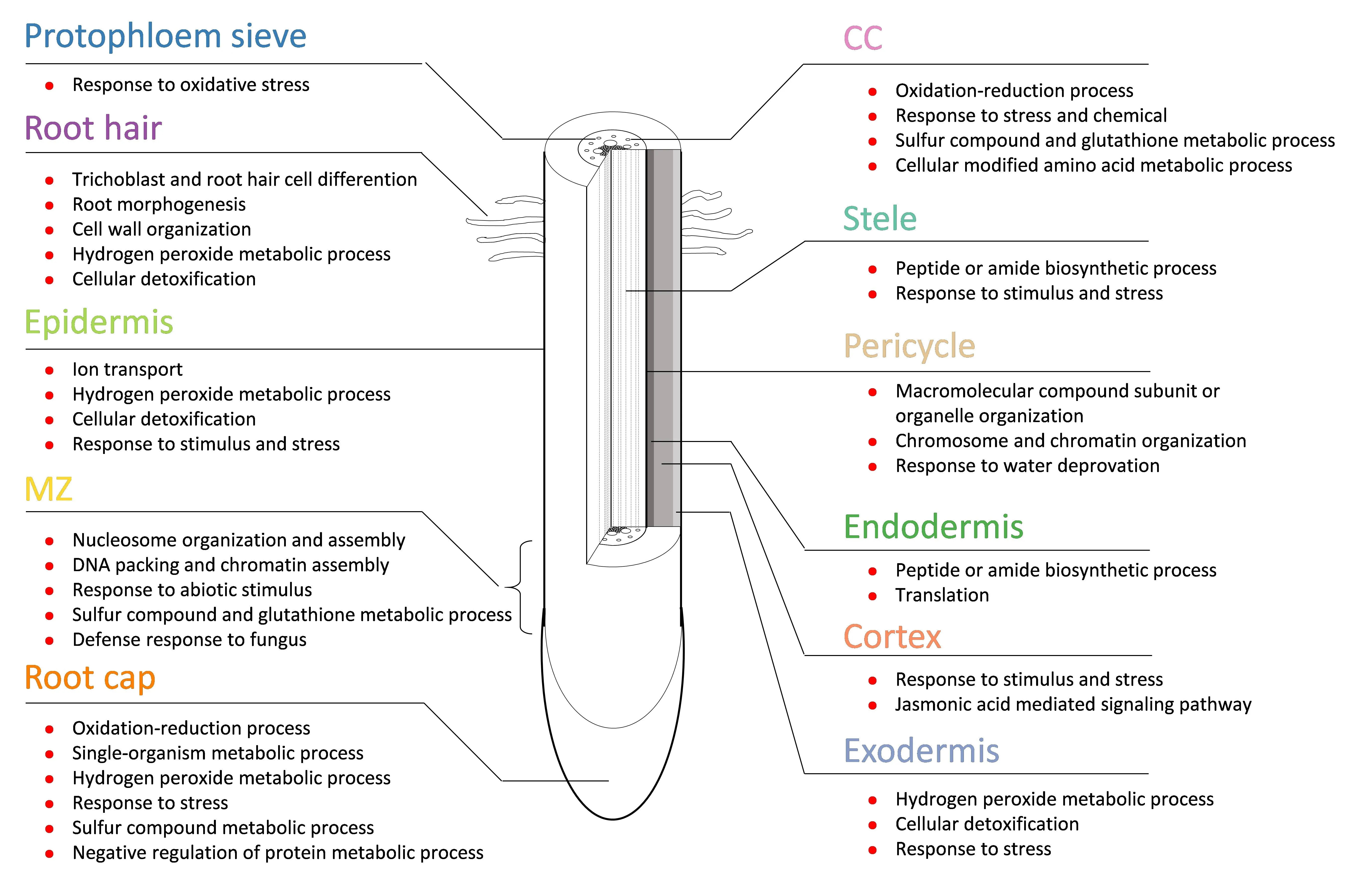
Fig.2.Schematic of root tip structure and GO function of marker genes of each cell type of maize root tips.
Transcription factors are critical regulators of organism growth and development.A total of 70 cell type-specific marker genes encoded transcription factors(TFs),and could be grouped into 15 families(Fig.S7).The ZIM and NAC families were predominant in maize root tips,containing respectively 15 and 14 marker genes.There were 33 TFs preferentially expressed in MZ cells.
3.3.Identification of cell type-specific nitrate-responsive genes
To characterize the response to nitrate in maize root at the single-cell level,single-cell transcriptomes of maize root growing in culture solutions with(nitrate+)or without nitrate(nitrate-)were profiled.The numbers and proportions of cell types of roots were similar between nitrate+and nitrate-(Table S4),and the correlations between nitrate+and nitrate-were high across all cell types except root cap,a finding attributed to a low number of root cap cells(17 cells for nitrate+and 2 cells for nitrate-)leading to underrepresentation(Fig.3B).To identify cell type-specific genes involved in the response to nitrate,cells of each cell types were split into two putative clusters according to the treatments.DEGs differentially expressed between putative clusters were then assigned as cell type-specific nitrate-response genes.A total of 85 DEGs were identified,with 48 up-and 37 down-regulated genes(Fig.3A-left;DataSet 5).Compared with the bulked RNA-seq,63 DEGs derived from scRNA-seq were collectively identified as nitrate-response genes by bulked RNA-seq(DataSet 5 and DataSet 6).The 39 DEGs identified in the cortex were associated with abiotic stimulus,hormone,and signal transduction by GO enrichment analysis(Fig.3A,right).Epidermis-specific marker genes were associated with ion transport and metabolism as shown by GO enrichment analysis.Two nitrate metabolism-associated genes,ZmNAR2.1(nitrate transporter/Zm00001d017095)and ZmGS2(glutamine synthetase 2/Zm00001d026501),were up-regulated in the epidermis of nitrate+roots as well as in root hair cells(Fig.3C-E).In MZ cells of maize roots,ZmNIR1(nitrite reductase1/Zm00001d018161)was also induced by nitrate(Fig.3C,F).ZmGS6(glutamine synthetase6/Zm00001d028260)was significantly induced in the cortex of roots grown in nitrate+media(Fig.S8AC;DataSet 5).The expressions of two NADPH synthesis related genes,which are essential for nitrate reduction,were increased in the epidermis of roots grown in nitrate+medium(Fig.S8D-I)[65,66].
3.4.Pseudotime analysis of major root cell-types
The root tip cell types showed a dynamic development process at the seedling stage,arising from a pool of undifferentiated apical meristematic cells.The differentiation was progressive and followed a continuum rather than discrete changes.Cells in different developmental states expressed different sets of genes,and scRNAseq enabled the capture of cells in both terminal and intermediate developmental states and made it possible to explore the continuous differentiation trajectory of a developmental process.The UMAP dimension reduction algorithm captures more of the global and topological structure of the datasets than others[67],including the spatial position of individual cells.Positional information is especially important for plant development because it is associated with cell differentiation and cell fate determination.We tried to assess the pseudotime for cell partition 1 with Monocle3 R package(Fig.4A),and obtained a continuous pseudotime series with the beginning at MZ cells(Figs.4B,S9).Two major putative developmental trajectories,the stele and cortex trajectories,were plotted manually,defining the two probable fates of MZ cells(Fig.4B).The pseudotime of cells was increased along with two trajectories.The cortex trajectory included the cortex and exodermis,but not endodermis.The stele trajectory included mainly stele,protophloem sieve,CC cells,and pericycle.
Identification of genes whose expression changes over pseudotime is an important objectives of developmental trajectory analysis.Knowing the order in which gene expression turns on or off can inform novel models of development.By spatial autocorrelation analysis,173 putative trajectory-dependent genes were identified at a cutoff of morans_I>0.3,and the expression of these genes changed with pseudotime or putative trajectories(Fig.S10;Data-Set 7).For example,the expression pattern of Zm00001d026163(encoding an extensin-like protein/RGG144)was not homogeneous in cortical cells,being lower in the cells near the base of the putative trajectory and gradually increasing along the putative trajectory curve(Fig.4C).
To examine the developmental trajectory of epidermal cells(root hair and epidermis cells),the slingshot algorithm[47]was adopted,and two branches with a common starting point were identified(Fig.4D-F).These cells could be further divided into four subgroups(Fig.4E).The putative trajectory started with a subset of the epidermis cells(subgroups 1 and 2),and the trajectory gradually ended with two differentiated cell types:the root hair cells(branch 1)and the remaining epidermis cells(branch 2)(Fig.4E).Marker genes for epidermis and root hair displayed specific expression in each cell type(Fig.4G-I).To character the gene expression pattern across the trajectory,the 100 most important genes of the trajectory were used for hierarchical clustering and fell into two major gene clusters.The cells of the two branches showed distinct gene expression patterns(Fig.4J;DataSet 8).Although part of the genes(at the bottom)of gene cluster1 tended to be highly expressed in branch2,the overall expression pattern did not exhibit branch specificity.Gene cluster 2 contained 44 gene members,and these genes were preferentially expressed in branch 1,which consisted of root hair cells.Not surprisingly,these genes were enriched in GO terms associated with trichoblast differentiation and plant-type cell wall organization(Fig.4J,right).These findings confirmed that root hair cells were differentiated from epidermis cells and reflected transcriptional rewiring during root epidermal cell differentiation,which recapitulated root hair development.
3.5.Root cell-types and cell-type-specific genes conserved between maize and rice
To investigate the similarities and differences between maize and rice roots,scRNA-seq profiles of maize and rice were compared.First,we obtained three published rice root scRNA-seq datasets from 9311,Nipponbare(Nip)and ZH11[16,25],and defined a set of one-to-one homologous gene pairs across maize and rice according to MaizeGDB.Then,7996 orthologous genes pairs were used for integration and further analysis(DataSet 1).Second,scRNA-seq datasets of rice and maize were integrated.Dimension reduction and cell clustering were performed(Fig.5A).In total,22 cell clusters were identified at a resolution of 0.6(Fig.5B).Because the cell types of rice had been annotated by each author,we could assign cell types to each cell cluster directly(Fig.5D).Conserved cell types were determined based on the UMAP coordinates and Pearson’s correlations between maize and rice(Fig.S11).A relatively high degree of similarity was identified in the root hair,endodermis,phloem,and exodermis.In particular,root hair cells from maize and rice showed relatively high correlations,and clustered into same cluster(cluster 14).Endodermis cells from maize and rice showed high correlations and similarity in their position in UMAP space[16],while endodermis cells from ZH11 had a slightly different position,although they showed a high correlation with the corresponding cell type from 9311 and Nip(Fig.S11).For phloem cells from maize(protophloem sieve)and rice(phloem)of ZH11,the Pearson’s correlation coefficients of gene expression were high and the positions of cells in UMAP space almost completely coincided with each other(Figs.5D,S11).The exodermis of maize showed a high correlation with Epidermis_Exodermis_S clerenchyma and exodermis of rice,and belonged to three clusters(0,12 and 6),and the positions of exodermis cells from maize and rice(ZH11)were in close proximity to each other.Spatial proximity often implies functional similarity;for example,the exodermis of maize showed a high correlation with the cortex of rice from ZH11(Fig.S11),suggesting that these cell types are functionally similar across species.
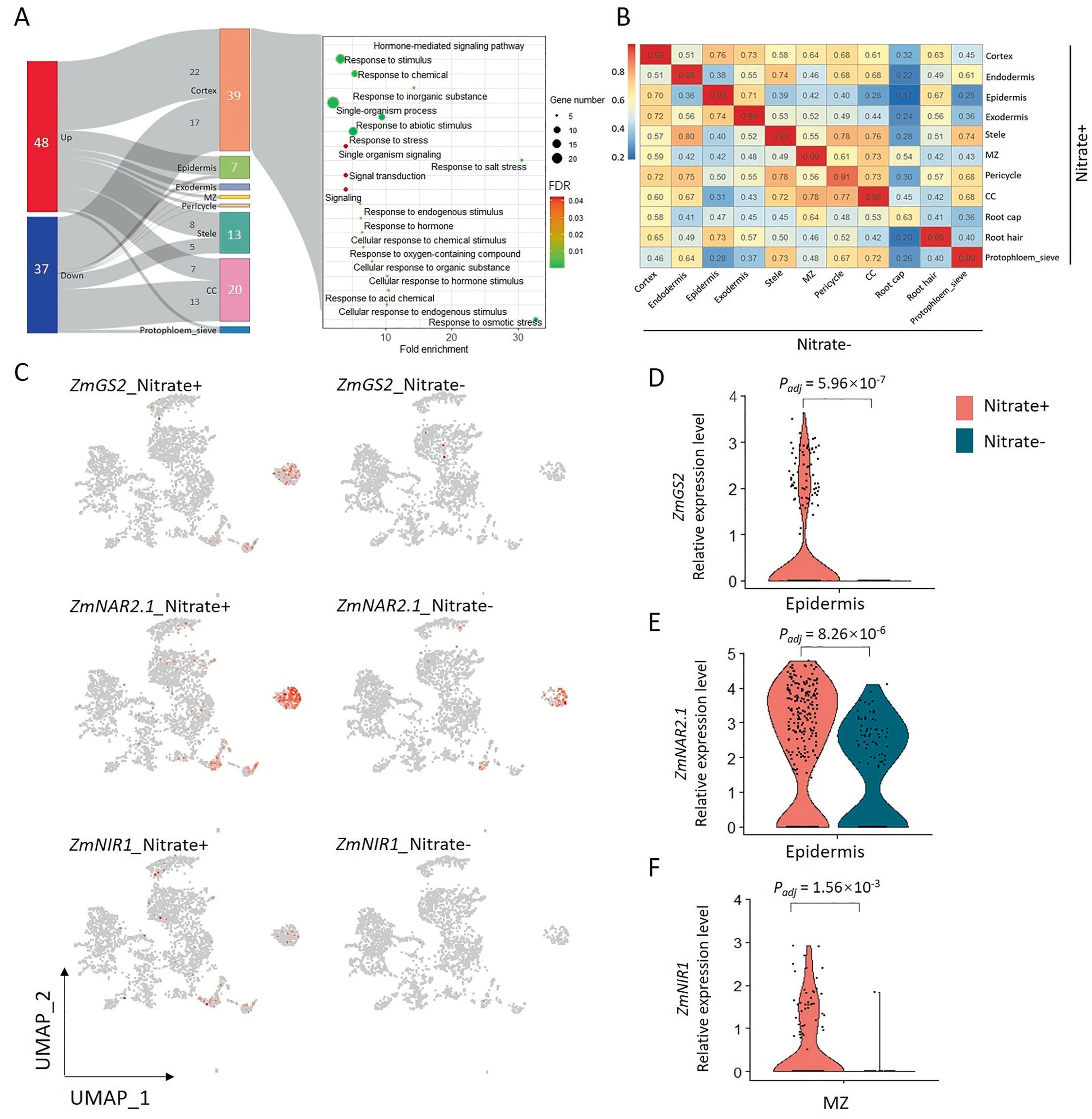
Fig.3.Cell type-specific nitrate-responsive genes.(A)Sankey diagram of cell type specific genes response to nitrate(left),and GO enrichment analysis of cortex cell-specific nitrate-responsive genes(right).(B)Heat map showing correlations between gene expression levels of cell types from nitrate+and nitrate-.(C)Scatter plot for three nitrate metabolism genes.(D-F)Violin plot showing expression differences for gln1(D),npf5(E),and nii1(F).Padj was calculated with a likelihood-ratio test(bimod).
Three conserved cell types:root hair,endodermis and phloem,were used to identify expressed genes of cell types conserved in maize and rice.Root hair cells of the five scRNA-seq datasets of maize and rice shared high transcriptome similarities.For root hair cells,gene clustering analyses revealed a total of 58 core orthologous genes(conserved genes)that were highly expressed in root hairs of both species,and GO enrichment analysis showed that these orthologous genes of maize were involved in root hair and epidermal cell development(Figs.5C,S12;DataSet 9).216 orthologous genes showed high expression in endodermis cell of both maize and rice(Nip and 9311)(Fig.S13A,left;DataSet 9),GO enrichment analysis revealed that these genes were involved in biogenesis of ribosomes and macromolecules(Fig.S13A,right).The orthologous pair ZmCASP1 and OsCASP3 showed endodermisspecific expression(Fig.S13B).For phloem cells,80 genes showed constitutive expression in maize and rice(ZH11)(Fig.S14;DataSet 9),and these genes were not enriched in any GO category.Zm00001d049606(used for FISH assay)and Os08g0138600 were constitutively expressed in phloem cells both in maize and rice.
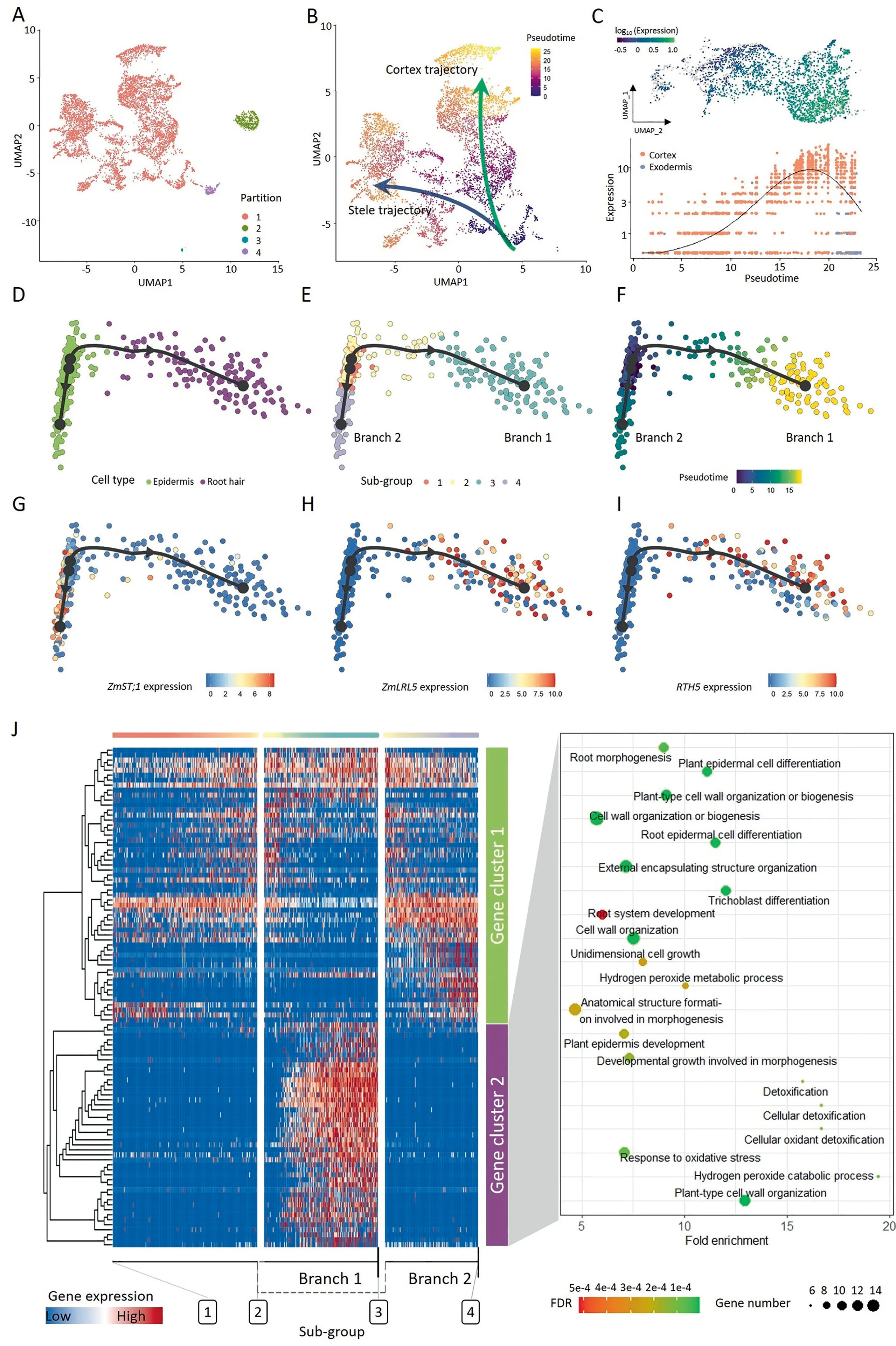
Fig.4.Pseudotime analysis and developmental trajectory of root tip cells.(A)Four cell partitions(superclusters)were identified in UMAP embedding.(B)Pseudotime and two major developmental trajectories were shown in two-dimensional UMAP space.(C)Expression of Zm00001d026163(RGG144),a trajectory-dependent gene,along with psudeotime in cortex cells.(D-F)Epidermal cells projected on two dimensions used the slingshot algorithm,cell types(D),sub-groups(E),and pseudotime(F).(G-I)Expression pattern of marker genes of epidermis(ZmST1;1)and root hair(ZmLRL5 and rht5).(J)Heat map showing the expression of 100 most important genes on two branches across pseudotime.All genes were grouped into two clusters with distinct expression patterns.Each row represents one gene.Gene functions revealed by GO enrichment analysis for gene cluster 2 are shown at right.
4.Discussion
4.1.scRNAseq is suitable for cell type identification in maize
The root of maize comprises plentiful heterogeneous cell types.In this study,we profiled nearly 7000 transcriptomes of maize primary root tip cells at the seedling stage and identified 11 major cell types or tissues,although we could not character all sub-cell-types(such as xylem)for stele and assign a cluster to QC cells,owing to their similarity in gene expression or rarity in number[17,21].We found cell-type assignment based on the expression of marker genes or transcriptome comparison to be difficult,although the cell types of root cap,root hair,and endodermis were relatively easily assigned based on the expression of marker genes.For example,ZmRCP1 and ZmRCP2 were highly abundant in the root cap[59],and RTH5,ZmLRL5 and RTH6 were expressed exclusively in root hair[54-56].Five CASP-family genes of maize,orthologs of Arabidopsis genes,were predominant in the endodermis(Fig.1F)[17,61].For comparison of the transcriptome,only the pericycle and root hair showed a relatively high and acceptable correlation coefficient(Fig.1G)[5,10].Despite the background noise in images[44],the mRNA FISH assay proved to be a useful tool for cell type annotation,and may in future allow identification of new cell types or sub-cell types(Fig.S3).Although it was not sequenced to saturation,our expression data also support the view that cell types were readily distinguishable[22],and allowed characterizing the cell types of maize primary roots.
4.2.Application of developmental trajectory analysis in recapitulating root development
Developmental trajectory analysis of cells revealed that almost all cell types of roots were derived from a common cell cluster(MZ),which contained meristematic cells,and two major putative trajectories(putative cortex trajectory and putative stele trajectory)were identified(Fig.4).In the putative stele trajectory,the phloem was assigned,and xylem cells might be mixed in the stele cells,a speculation that invites further investigation.In the putative cortex trajectory, the expression of RGG144(Zm00001d026163)was increased with pseudotime in cortex cells(Fig.4C).RGG144 was expressed mainly in the region far from the root cap[68],suggesting that the cell’s position in pseudotime provides spatial information.These results showed that it was feasible to reconstruct the developmental trajectory in UMAP space by appropriate methods.
Root hairs are long tubular outgrowths that form on the surface of specialized epidermal cells.Their development occurs in three phases:cell fate specification,initiation,and subsequent tip growth[69].Pseudotime analysis of epidermal cells(root hair and epidermis cells)revealed that the root hair differentiated from a subset of epidermis cells(Fig.4D-J),a finding consistent with the results of single-cell sequencing assays for transposase-accessible chromatin(scATAT-seq)in maize and scRNA-seq in rice[16,17,25].Gene expression analyses indicated that the expressions of root hair development-associated genes were specific and higher than those of other epidermal cells,suggesting the potential for reconstructing developmental trajectories by analyzing the transcriptome of each cell and identifying developmentdependent genes.
4.3.The potential of scRNA-seq for identification of cell type-specific response genes
By comparing cell types from roots grown on nitrate+or nitrate-media,we discovered that nitrate does not distinctly alter the cell type,even the number of each cell type(Table S2),but does show substantial differences in the numbers of nitrate-response genes identified in eight cell types,especially in cortex and CC(DataSet 5;Fig.3).Several genes involved in nitrate absorption and assimilation were induced specifically in epidermal and MZ cells.The expression of ZmGS2(glutamine synthetase 2/Zm00001d026501)and ZmNAR2.1(high-affinity nitrate transporter/Zm00001d017095)were induced in epidermis cells of roots grown on nitrate+media(Fig.3).It was reported[70]that the expressions of both ZmGS2 and ZmNAR2.1 were induced specifically by nitrate instead of by other nitrogen forms.We also identified two genes(ZmGS6 and ZmNIR1)and two NADPH synthesis-associated genes that were more highly expressed in roots grown on nitrate+media,and function inassimilation[65,66].Although these nitrate-induced genes have been shown to be involved in nitrate absorption and reduction,we clarified their expression patterns at the cell-type level.These results pave the way for new applications in biotechnology and empower the generation of de novo phenotypic variation in crops.Overexpression of transgenes in all tissues of crops often leads to unpredictable phenotypic changes.We identified a series of cell type-specific nitrate-response genes that could serve as targets to tailor the expression of genes in given tissues of transgenic plants.Using this strategy,researchers could more accurately regulate gene expression and control phenotypic variation.Thus,the influence of exogenous gene expression on receptors can be reduced to the greatest extent and can reduce some of the risks of genetically modified(GM)crops.
4.4.Root hair cell-specific genes conserved between maize and rice
LRL5 and OsRHL1,an orthologous gene pair,function as key regulators of root hair development in maize and rice[56,71]OsSNDP1,the ortholog of Zm00001d016601,encodes a Sec14-nodulin domain-containing protein.Huang et al.[72]reported that a single amino acid substitution of OsSNDP1 led to aberrant morphogenesis in root hair elongation and the maturation zones of rice,although many epidermal cells produced dome-shaped root hairs,indicating that root hair elongation ceased at an early stage.Zm00001d021489 encodes rop guanine nucleotide exchange factor(RopGEF).The root hair of the silenced MtRopGEF2 line was only about one-third the length of the wild type(WT)line in Medicago truncatula[73].It has been shown[74-76]that RopGEF interacts with RAC/ROPs and then regulates root hair development.These results show the functional similarity of conserved orthologous genes between maize and rice,and provide a new potential method for discovery of cell type-or tissue-specific genes.The conserved and divergent orthologous genes may contribute to the similarities and differences in functions of root cell types among species,though these differences might also have been caused by the sampling length of root tips.
In summary,we identified and characterized almost all major cell types of maize root tips,as well as a series of cell typespecific nitrate-response genes.Although not all cell types were captured,our results provide a valuable resource for investigating cell-type differentiation and the response to nitrate stimuli.Comparison of scRNA-seq data from maize and rice revealed evolutionarily conserved cell type and genes.The genes characterized in this study could serve as targets for gene transfer and further genetic analyses,and help researchers to accurately control gene expression and phenotypic variation in specific cell types or tissues.
Data availability
Single-cell RNA-seq and bulked RNA-seq data are available at NCBI under GEO accession number GSE183171.
CRediT authorship contribution statement
Xuhui Li:Conceptualization,Writing-original draft,Formal analysis,Visualization.Xiangbo Zhang:Methodology,Data curation.Shuai Gao:Methodology,Formal analysis.Fangqing Cui:Methodology.Weiwei Chen:Methodology,Writing-review &editing.Lina Fan:Methodology,Validation.Yongwen Qi:Conceptualization,Funding acquisition,Writing-review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This research was financially supported by the Special Project of Guangdong Academy of Sciences,China(2020GDASYL-20200103073),the Special Project for Research and Development in Key areas of Guangdong Province(2019B020238001),and the National Natural Science Foundation of China(32072027).
Appendix A.Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2022.02.004.

- The Crop Journal的其它文章
- Profiling seed soluble sugar compositions in 1164 Chinese soybean accessions from major growing ecoregions
- Strigolactones interact with other phytohormones to modulate plant root growth and development
- Wheat breeding for Hessian fly resistance at ICARDA
- Amino acid permease 6 regulates grain protein content in maize
- Comparative genomic analyses reveal cis-regulatory divergence after polyploidization in cotton
- Dynamic and adaptive membrane lipid remodeling in leaves of sorghum under salt stress