
Eight hub genes as potential biomarkers for breast cancer diagnosis and prognosis:A TCGA-based study
2022-11-29 08:49:14NanLiuGuoDuoZhangPingBaiLiSuHaoTianMiaoHe
Nan Liu, Guo-Duo Zhang, Ping Bai, Li Su, Hao Tian, Miao He
Nan Liu, Guo-Duo Zhang, Ping Bai, Li Su, Miao He, Department of Hematology and Oncology,Chongqing Traditional Chinese Medicine Hospital, Chengdu University of Traditional Chinese Medicine, Chongqing 400011, China
Hao Tian, Department of Breast and Thyroid Surgery, Southwest Hospital, Army Medical University, Chongqing 400038, China
Abstract BACKGROUND Breast cancer (BC) is the most common malignant tumor in women.AIM To investigate BC-associated hub genes to obtain a better understanding of BC tumorigenesis.METHODS In total, 1203 BC samples were downloaded from The Cancer Genome Atlas database, which included 113 normal samples and 1090 tumor samples. The limma package of R software was used to analyze the differentially expressed genes (DEGs) in tumor tissues compared with normal tissues. The cluster Profiler package was used to perform Kyoto Encyclopedia of Genes and Genomes (KEGG)enrichment analysis of upregulated and downregulated genes. Univariate Cox regression was conducted to explore the DEGs with statistical significance.Protein-protein interaction (PPI) network analysis was employed to investigate the hub genes using the CytoHubba plug-in of Cytoscape software. Survival analyses of the hub genes were carried out using the Kaplan-Meier method. The expression level of these hub genes was validated in the Gene Expression Profiling Interactive Analysis database and Human Protein Atlas database.RESULTS A total of 1317 DEGs (fold change > 2; P < 0.01) were confirmed through bioinformatics analysis, which included 744 upregulated and 573 downregulated genes in BC samples. KEGG enrichment analysis indicated that the upregulated genes were mainly enriched in the cytokine-cytokine receptor interaction, cell cycle, and the p53 signaling pathway (P < 0.01); and the downregulated genes were mainly enriched in the cytokine-cytokine receptor interaction, peroxisome proliferator-activated receptor signaling pathway, and AMP-activated protein kinase signaling pathway (P < 0.01).CONCLUSION In view of the results of PPI analysis, which were verified by survival and expression analyses, we conclude that MAD2L1, PLK1, SAA1, CCNB1, SHCBP1, KIF4A, ANLN, and ERCC6L may act as biomarkers for the diagnosis and prognosis in BC patients.
Key Words: Breast cancer; Bioinformatics; Hub gene; The Cancer Genome Atlas; Protein-protein interaction
INTRODUCTION
Breast cancer (BC) is the most common malignant tumor in women. In 2019, 268600 new BC patients and 41760 new BC deaths were reported, accounting for 30% of all new cancer cases and 15% of cancerrelated deaths, respectively. The mortality of BC is second only to lung cancer[1]. In recent years, BC outcome has significantly improved and treatment strategies such as surgery, chemotherapy,radiotherapy, endocrine therapy, and targeted therapy have achieved fine clinical benefits[2], whereas patients with distant metastases are almost incurable[3]. In addition, even after resection of the primary tumor, 30% of early BC is prone to recurrence in distant organs[4]. In clinical practice, the treatment and prognosis of different molecular subtypes of BC are significantly different: estrogen receptor-positive(ER+) patients prefer endocrine therapy, human epidermal growth factor receptor 2-positive (HER2+)patients prefer targeted therapy, and poorly differentiated tumors are usually associated with a poor prognosis[5-7].
Recent studies have found that the occurrence and development of BC are related to many molecular markers. For example, the expression of cluster of differentiation 82 is significantly decreased in BC and is associated with disease progression and metastasis[8]. In addition, a study on triple-negative BC suggested that multiple long noncoding RNAs are associated with prognosis, including MAGI2-AS3,GGTA1P, NAP1L2, CRABP2, SYNPO2, MKI67, and COL4A6[9]. Advances in microarray and highthroughput sequencing technology provide strong support for the development of more reliable prognostic markers[10,11]. Genome wide expression profiling can reveal molecular changes in the process of tumorigenesis and development, and has proven to be an efficient method to identify key genes[12]. Therefore, it is particularly important to explore more sensitive and specific biomarkers to further understand the pathogenesis of BC and the choice of treatment strategies.
This public database-based study explored potential hub genes in the occurrence and development of BC through bioinformatics analysis of the gene expression profile and clinical characteristics of BC, in order to provide new biological targets and directions for the clinical diagnosis and treatment of BC.
MATERIALS AND METHODS
Data sources and processing
The Cancer Genome Atlas (TCGA) database is a cancer research project established by the. National Cancer Institute and National Human Genome Research Institute. It aims to understand the mechanism of carcinogenesis and development of cancer cells and develop new diagnosis and treatment methods by collecting various types of cancer-related omics data. In this study, 1203 breast samples (fragments per kilobase million [FPKM] format) were downloaded from TCGA database (https://portal.gdc.cancer.gov/), including 1090 tumor samples and 113 normal samples. For a more accurate comparison of gene expression, FPKM data were converted to transcripts per million (TPM). At the same time, 1097 tumor samples containing clinical information were downloaded, and the data that did not match the expression samples were excluded. The remaining 1089 tumor samples were included in the univariate Cox regression analysis. Overall survival (OS) was taken as the endpoint event, and gene expression in TPM format was converted to log2(x + 1).
DEG acquisition
Limma package of R software (version 3.6.3) was employed for differential gene analysis[13], using the adjusted P-value (adj P-value) to avoid false-positive results. The inclusion criteria of DEGs were: | log2 fold change (FC) | > 2 and adjusted P < 0.01. The ggplot2 package of R software was used to generate a volcano plot to visualize these differential genes.
Functional enrichment analysis
DEGs were converted into gene ID through org.Hs.eg.db package of R software, and then Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was carried out by R software's clusterProfiler and enrichlot program package. ggplot2 program package was used to display the top 10 enrichment items, and adjusted P < 0.05 was considered statistically significant.
Univariate Cox regression analysis
The survival package of R software was used to carry out univariate Cox regression analysis on 1089 BC samples with survival information. The median value of expression was set as the cut-off point between the high expression and low expression groups, and differential genes related to prognosis were obtained for subsequent analysis. P < 0.05 was considered statistically significant.
Construction of PPI
The STRING database (https://string-db.org/) is a search tool for searching interacting genes, which aims to construct protein-protein interaction (PPI) networks of different genes based on known and predicted PPIs, and analyze the proteins that interact with each other[14]. Based on the online tool STRING, PPI of prognosis-related DEGs was constructed, and the confidence score was ≥ 0.4. Then the PPI network was visualized by Cytoscape software (version 3.7.2). In addition, using the CytoHubba plug-in of Cytoscape software to calculate the gene degree through the “degree” method, the top 10 genes were taken as the hub genes for subsequent analysis and verification.
Survival analysis of hub genes
The Kaplan-Meier plotter (http://kmplot.com/analysis/) can use 18674 cancer samples to evaluate the impact of 54675 genes on survival[15]. These studies included recurrence-free survival and OS information of 5143 cases of BC, 1816 cases of ovarian cancer, 2437 cases of lung cancer, 1065 cases of gastric cancer, and 364 cases of liver cancer, which are mainly based on Gene Expression Omnibus,TCGA, and European Genome-phenome Archive databases. The role of the tool is to benefit patients in clinical decision making, health care policy, and resource allocation through meta-analysis of biomarker assessment[16]. In this study, we analyzed the OS rate of 10 hub genes in BC using the Kaplan-Meier plotter. According to the median expression of each hub gene in Kaplan-Meier plotter, the patients were divided into two groups to present the difference in survival probability between the high expression group and the low expression group. A total of 14 datasets were enrolled in our analysis according to the Kaplan-Meier web tool and detailed retrospective clinical information in http://kmplot.com/analysis/. P < 0.05 was considered statistically significant.
To further investigate the prognostic value of the hub genes selected above, we performed the logrank test on these hub genes in molecular subtypes of BC based on TCGA cohort. Through the PAM50 algorithm, TCGA cohort was separated into five major subtypes: luminal A, luminal B, HER2 enriched,basal-like, and normal-like. This method was completed through utilizing the “genefu” R package according to detailed operation protocol.
Expression analysis of hub genes
The Gene Expression Profiling Interactive Analysis (GEPIA) database was employed to verify the mRNA expression levels of 10 hub genes in normal breast tissues and cancer tissues. GEPIA database contains data from 9736 tumor samples and 8587 normal samples, which were used to display the mRNA expression levels of each key gene in cancer and non-cancer tissues[17]. The protein expression levels of 10 hub genes in human normal tissues and BC tissues were analyzed using the human protein atlas database (HPA), which contains immunohistochemical expression data covering about 20 of the most common types of cancer[18].
RESULTS
Identification and functional analysis of DEGs
After DEG analysis of 113 normal breast samples and 1090 BC samples, we found that there were 1317 DEGs, of which 744 were upregulated and 573 were downregulated in BC. As shown in Figure 1A, red represents high expression and blue represents low expression. At the same time, the volcano plot was used to present the distribution of DEGs (Figure 1B), the red dots represent upregulated genes and the blue dots represent downregulated genes.
To further understand the biological function of these 1317 DEGs, the clusterProfiler and enrichplot packages of R software were used to perform KEGG enrichment analysis on these DEGs. The enrichment analysis results of upregulated genes and downregulated genes are shown in Figure 1C and D, respectively. The top 10 upregulated genes were the cytokine-cytokine receptor interaction,neuroactive ligand-receptor interaction, cell cycle, oocyte meiosis, interleukin 17 signaling pathway,cellular senescence, progesterone-mediated oocyte maturation, p53 signaling pathway, nicotineaddiction, and bladder cancer. The 10 ten downregulated genes were the cytokine-cytokine receptor interaction, peroxisome proliferator-activated receptor (PPAR) signaling pathway, AMP-activated protein kinase (AMPK) signaling pathway, retinol metabolism, tyrosine metabolism, adipocytokine signaling pathway, drug metabolism - cytochrome p450, ATP-binding cassette transporters, regulation of lipolysis in adipocytes, and fatty acid degradation.
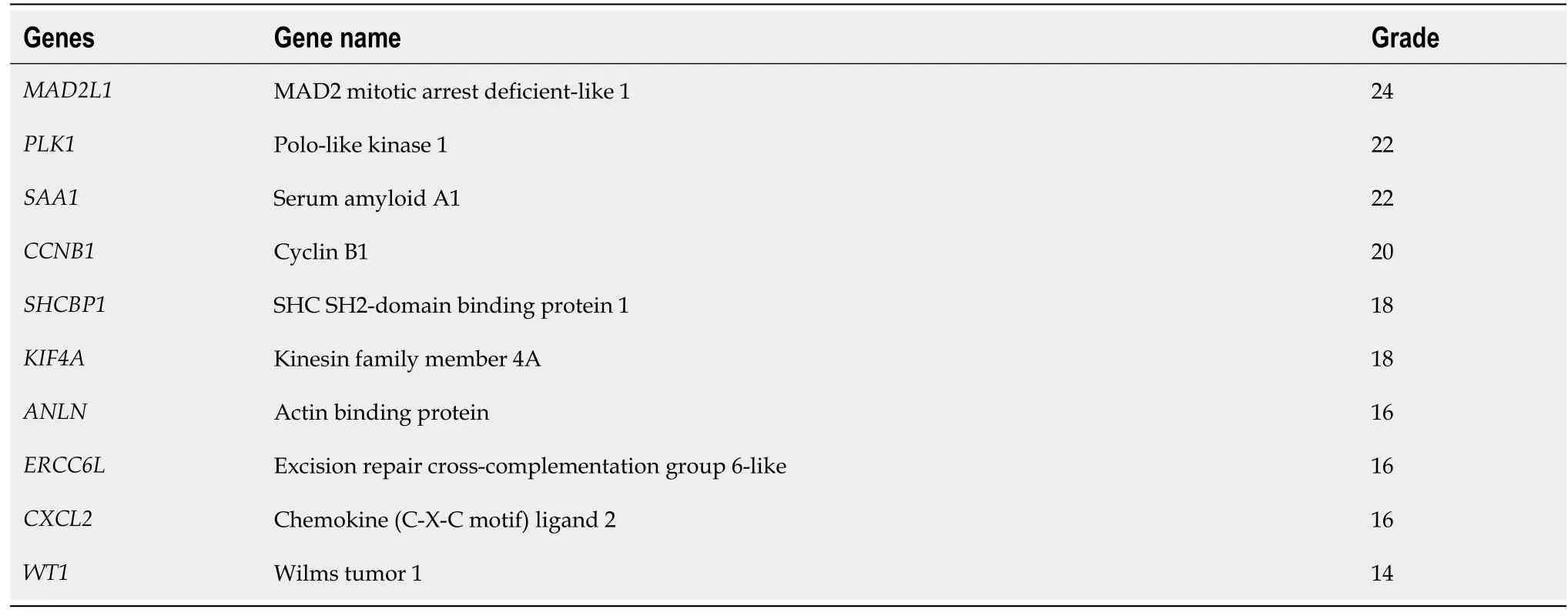
Table 1 Summary of the top 10 hub genes according to their grade
Screening of hub genes
To screen the DEGs related to the prognosis of BC, we used the survival package of R software to perform univariate Cox regression analysis on 1317 DEGs, and found that the prognosis of 165 genes was statistically significant (Supplementary Table 1). As shown in Figure 2, further analysis of the PPI of these 165 genes revealed that there were a total of 164 nodes and 156 interactions (edges), and the confidence score adopted default value ≥ 0.4. The CytoHubba algorithm of Cytoscape software was used to calculate the degree score of each node. The top 10 genes were MAD2L1, PLK1, SAA1, CCNB1,SHCBP1, KIF4A, ANLN, ERCC6L, CXCL2, and WT1 (Figure 3). The upregulated genes were represented by red and round nodes, and the downregulated genes were represented by blue and diamond nodes. The node size represented the level, and most of the hub genes were upregulated DEGs. Gene annotation and grade scores are shown in Table 1.
Survival analysis of hub genes
Kaplan-Meier plotter was used to explore the prognostic value of 10 hub genes in BC. The results showed that, except for CXCL2 [hazard ratio (HR) 0.86 (0.69-1.07); P = 0.170] and WT1 [HR 1.03 (0.83-1.28); P = 0.760], the highly expressed MAD2L1 [HR 2.02 (1.62-2.51); P = 1.8e-10], PLK1 [HR 1.42 (1.15-1.76); P = 0.0012], CCNB1 [HR 1.42 (1.04-1.94); P = 0.028], SHCBP1 [HR 1.76 (1.42-2.19); P = 2.1 e-07],KIF4A [HR 1.8 (1.44-2.23); P = 8.8e-08], ANLN [HR 1.48 (1.08-2.03); P = 0.014], and ERCC6L [HR 1.68(1.35-2.09); P = 2e-06] were related to the poor OS rate of BC patients. By contrast, the high expression of SAA1 [HR 0.71 (0.57-0.88); P = 0.018] was associated with a better OS rate for BC patients (Figure 4).
We also conducted the survival analysis of these 10 hub genes in TCGA molecular subtypes. As a result, TCGA cohort was successfully divided into five subtypes based PAM50 identifier: 563 of luminal A, 215 of luminal B, 82 of HER2-enriched, 189 of basal-like, and 39 of normal-like. Then survival analysis of these 10 genes was performed in each subtype group. The results indicated that CXCL2 (HR = 0.45; P< 0.05) and SAA1 (HR = 0.53; P < 0.05) were protective factors in the luminal A subtype (Figure 5).ANLN (HR = 2.12; P < 0.05), ERCC6L (HR = 3.04; P < 0.05), KIF4A (HR = 2.50; P < 0.05), PLK1 (HR = 2.40;P < 0.05), and SHCBP1 (HR = 2.42; P < 0.05) were hazard factors in luminal B subtype, whereas the CXCL2 (HR = 0.45; P < 0.05) showed protective effects. Finally, KIF4A (HR = 4.31; P < 0.05) acted as a risk factor in HER2-enriched patients and CXCL2 played a satisfactory role among basal-like patients(HR = 0.46; P < 0.05).
Expression analysis of hub genes
To verify the expression differences of key genes in BC, GEPIA was employed to analyze the mRNA expression levels of MAD2L1, PLK1, SAA1, CCNB1, SHCBP1, KIF4A, ANLN, ERCC6L, CXCL2, and WT1 between BC and non-cancerous tissues (Figure 5). Compared with non-cancerous tissues, MAD2L1(Figure 5A), PLK1 (Figure 5B), CCNB1 (Figure 5D), SHCBP1 (Figure 5E), KIF4A (Figure 5F), ANLN(Figure 5G), and ERCC6L (Figure 5H) in BC tissues were significantly upregulated (P < 0.01); SAA1(Figure 5C) and CXCL2 (Figure 5I) were significantly downregulated in BC (P < 0.01); and WT1(Figure 5J) tended to increase in BC tissues. After verifying the mRNA expression level of hub genes, we used the HPA database to verify the protein expression level of these hub genes in BC. It is worth noting that MAD2L1 (Figure 6A), PLK1 (Figure 6B), CCNB1 (Figure 6C), SHCBP1 (Figure 6D), ANLN(Figure 6F), ERCC6L (Figure 6G), and WT1 (Figure 6H) were not expressed in normal breast tissues, but expressed in different levels in BC tissues. KIF4A (Figure 6E) was moderately expressed in normal breast tissues and highly expressed in BC tissues. In short, the expression of hub genes was consistent with the results of differential analyses at both the mRNA and protein levels.
DISCUSSION
In this study, we used bioinformatics analysis to screen and verify potential biomarkers associated with BC. After comparing the gene expression matrix of breast tissue retrieved from TCGA database, 744 upregulated DEGs and 573 downregulated DEGs were successfully identified. Combined with the survival data, 165 prognostic-related DEGs were analyzed. According to PPI network analysis, the top 10 node genes were ranked: MAD2L1, PLK1, SAA1, CCNB1, SHCBP1, KIF4A, ANLN, ERCC6L, CXCL2,and WT1. After subsequent survival analysis and expression analysis verification, the expression and prognosis of MAD2L1, PLK1, SAA1, CCNB1, SHCBP1, KIF4A, ANLN, and ERCC6L in BC were finally confirmed. These eight hub genes may play a vital role in the occurrence and development of BC.
Among the 1317 identified DEGs, significant gene expression dysregulation was observed in the cell cycle, PPAR signaling pathway, and AMPK signaling pathway. Cell cycle is a highly conserved process in human evolution and is essential for the normal growth of cells. Abnormal cell cycle is a hallmark of human cancer[19]. Recent studies have also identified several genes related to the cell cycle, including CCNB1, ANLN, MAD2L1, and PLK1. For example, CCNB1 may be a biomarker for the prognosis of ER+BC patients and monitoring the efficacy of hormone therapy[20]. Recent studies have found that the occurrence and proliferation of gastric cancer cells induced by ISL1 is mediated by the expression and regulation of CCNB1, CCNB2, and C-MYC[21]. In addition, the high expression of ANLN in BC cell nuclei is significantly related to tumor tissue size, histopathological grade, high proliferation rate, and a worse prognosis[22]. MAD2L1 is a mitotic spindle checkpoint gene. In patients with primary BC,compared with patients with ER+, PR+ and low-grade tumors, patients with ER-, PR- and high-grade tumors have higher expression of MAD2L1, and high expression of MAD2L1 is associated with a poor OS[23]. PLK1 is a key oncogene that can regulate the transition of cells in the G2-M phase, thus promoting the growth and metastasis of tamoxifen resistant BC[24]. These studies are consistent with our current conclusion that CCNB1, ANLN, MAD2L1, and PLK1, as key genes, are overexpressed in BC tissues, and their overexpression is correlated with poor prognosis. Meanwhile, the PPAR signaling pathway may be an important predictor of BC response to neoadjuvant chemotherapy[25], and activation of the AMPK signaling pathway can inhibit the activity of the Wnt/β-catenin signaling pathway, thereby inhibiting the growth of BC cells[26]. These studies showed that the identified DEGs play a critical role in the occurrence and development of BC, and the hub genes among them may serve as prognostic markers and are worth further investigation.
With the exception of CCNB1, ANLN, MAD2L1, and PLK1, the gene combination model of CD74,MMP9, RPA3, and SHCBP1 in the tumor microenvironment (TME) can effectively predict the prognosis and disease risk of BC patients[27], while their potential mechanism remains unknown. In addition, the circKIF4A-miR-375-KIF4A axis can regulate the development of triple-negative BC through competing endogenous RNA, and circKIF4A can act as a prognostic biomarker and therapeutic target for triple
negative BC[28].
SAA1 is a serum amyloid protein family member that is highly expressed in non-small cell lung cancer, and is associated with a poor prognosis and tyrosine kinase inhibitors[29]. SAA1 has low expression in hepatocellular carcinoma, and the high expression of SAA1 is associated with a better prognosis[30]. To date, SAA1 has not been reported in BC, and the specific role and function of this gene in BC require further experimental exploration and clinical specimen verification. ERCC6L is a newly discovered DNA helicase. In the human BC cell line MDA-MB-231, exogenous interference with the expression of ERCC6L can inhibit the growth of BC cells[31]. However, its role and specific mechanism in clinical specimens are still unknown. The expression of ERCC6L is upregulated in clear cell renal cell carcinoma, and the highly expressed ERCC6L can promote the proliferation of clear cell renal cell carcinoma cells by regulating the mitogen-activated protein kinase signaling pathway[32]. In this study,we found that SAA1 and ERCC6L may be used as prognostic markers for BC, whereas there are few reports on these two genes, and further research is necessary.
In this study, we found that the differential expression of the eight hub genes are related to the occurrence and development of BC, and are significantly related to the OS rate, which indicate that these hub genes may be utilized as potential prognostic biomarkers and therapeutic targets for BC. This study had some limitations. First, due to the complexity of the dataset in the public database, it is difficult to consider some important confounding factors such as different ages, races, regions, and tumor stages when analyzing DEGs. Second, according to the results, seven key genes were upregulated in BC and one key gene was downregulated, but the mechanism of their differential expression is still unclear, and more studies are needed to confirm their biological basis. Finally, this study focused on the expression level and OS rate of the eight hub genes, and whether these key genes can be used as biomarkers and can improve the diagnostic accuracy and specificity of BC requires further research.
CONCLUSION
In conclusion, based on comprehensive bioinformatics analysis, this study identified 1317 DEGs related to the occurrence and development of BC, 165 DEGs related to prognosis, and 8 hub genes (MAD2L1,PLK1, SAA1, CCNB1, SHCBP1, KIF4A, ANLN and ERCC6L). Each of these eight hub genes has different expression levels in BC and is significantly related to prognosis. The results of this study indicate that studying these DEGs would help us have a deeper understanding of the molecular mechanisms of the pathogenesis and progression of BC. Moreover, these hub genes may serve as potential prognostic markers and therapeutic targets for BC, which provides a reference for more in-depth and extensive prospective clinical research.
ARTICLE HIGHLIGHTS
Research background
Breast cancer (BC) is the most common malignant tumor in women. In 2019, 268600 new BC patients and 41760 new BC deaths were reported, accounting for 30% of all new cancer cases and 15% of cancerrelated deaths. Therefore, it is particularly important to explore more sensitive and specific biomarkers for further understanding the pathogenesis of BC and the choice of treatment strategies.
Research motivation
Exploring more valuable therapeutic targets would be helpful in treating with high efficacy.
Research objectives
This study aimed to identify novel biomarkers for BC.
Research methods
The limma package of R software and clusterProfiler package were used to analyze the differentially expressed genes (DEGs) in tumor tissues compared with the normal tissues, respectively. The proteinprotein interaction network (PPI) analysis was used to investigate the hub-genes through cytohubba algorithm by the Cytoscape software. Survival analysis of the hub-genes were carried out through the Kaplan-Meier database. The expression level of these hub-genes was validated in the GEPIA database and the Human Protein Atlas database.
Research results
Upregulated genes mainly enriched in the cytokine-cytokine receptor interaction, cell cycle, and p53 signaling pathway (P < 0.01). The downregulated genes were mainly enriched in the cytokine-cytokine receptor interaction, peroxisome proliferator-activated receptor signaling pathway, and AMP-activated protein kinase signaling pathway (P < 0.01).
Research conclusions
MAD2L1, PLK1, SAA1, CCNB1, SHCBP1, KIF4A, ANLN, and ERCC6L may act as biomarkers for diagnosis and prognosis in BC patients.
Research perspectives
Proper validations must be made in future studies.
FOOTNOTES
Author contributions:Liu N performed the experiment and wrote the paper; Liu N, Zhang GD, and Bai P contributed to the bioinformatics analysis and figure preparation; Tian H and Su L modified the structure and language of the manuscript; He M and Tian H contributed to the conception and design of the study and the revisions of the manuscript; All authors have read and approved the final manuscript.
Institutional review board statement:Not applicable.
Conflict-of-interest statement:The authors have no conflicts of interest to declare.
Data sharing statement:No additional data are available.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORCID number:Nan Liu 0000-0003-1617-0138; Guo-Duo Zhang 0000-0002-2088-4590; Ping Bai 0000-0001-7863-452X; Li Su 0000-0001-9590-3402; Hao Tian 0000-0002-8606-6806; Miao He 0000-0002-4889-7959.
S-Editor:Gong ZM
L-Editor:Filipodia
P-Editor:Gong ZM
 World Journal of Clinical Oncology2022年8期
World Journal of Clinical Oncology2022年8期
- World Journal of Clinical Oncology的其它文章
- COVID-19 and oral cancer: Critical viewpoint
- Impact of cytochrome P450 2D6 polymorphisms on decision-making and clinical outcomes in adjuvant hormonal therapy for breast cancer
- Computer-aided clinical image analysis as a predictor of sentinel lymph node positivity in cutaneous melanoma
- Propensity-matched analysis of patients with intrahepatic cholangiocarcinoma or mixed hepatocellular-cholangiocarcinoma and hepatocellular carcinoma undergoing a liver transplant
- Potential of mRNA vaccines to become versatile cancer vaccines