
全自动膳食纤维仪测定食品中总膳食纤维的不确定度相关研究
2022-11-18 07:41刘艇飞肖晓峰
农产品加工 2022年19期
林 玲,刘艇飞,肖晓峰
(台州海关综合技术服务中心,浙江台州 317700)
膳食纤维[1]存于植物细胞壁中,是一种碳水化合物,膳食纤维=可溶性膳食纤维[2]+不可溶性膳食纤维,分为非水溶性和水溶性纤维两类。非水溶性纤维分为纤维素、半纤维素和木质素,主要来自根茎蔬菜和食物中的芹菜、果皮。非水溶性纤维可促进消化道中细菌排出的毒素,同时可降低罹患肠癌的风险[3]。水溶性膳食纤维[4]主要来自食物中的大麦、豆类、胡萝卜、柑橘、燕麦和燕麦糠等,水溶性膳食纤维可让血液中的血糖和胆固醇控制在理想的水平,还可以帮助糖尿病患者改善胰岛素和三酸甘油脂。饮食均衡摄取水溶性与非水溶性纤维可减缓消化速度和快速排泄胆固醇,有助于调节免疫系统功能,促进体内有毒重金属的排出[5]。因此,膳食纤维含量的测定是有必要的,通过全自动膳食纤维分析仪测定膳食纤维的含量可缩短复杂的中间处理环节,只需先将样品根据标准经过干燥、脱脂、脱糖等处理后按要求称入仪器即能快速完成分析,再通过后续的检测和计算得出数据,具有节省资源、检测快速的优点。但其测定结果的准确性又受到酶解过程及灰分含量、蛋白质含量等影响因素的影响,对上述因素做系统分析是有必要的,从而能够更加准确、高效测定食品中膳食纤维含量[6]。《GB 5009.88—2014》食品安全国家标准 食品中膳食纤维的测定[7],检测原理为干燥试样经过热稳定α -淀粉酶、蛋白酶和葡萄糖苷酶酶解消化以去除蛋白质和淀粉后,经过乙醇沉淀、抽滤,残渣用乙醇和丙酮洗涤,干燥称量,得到总膳食纤维残渣。扣除各类膳食纤维残渣中相应的蛋白质[8]、灰分[9]和试剂空白含量,即可计算出试样中总的膳食纤维含量[10]。通过测量不确度的评定,可以为测量结果质量的控制提供重要技术参考。
1 检测程序[9]
1.1 操作方法[10]
试样制备:试样处理根据水分含量、脂肪含量和糖含量进行适当的处理及干燥,并粉碎、混匀过筛。
脂肪含量<10%的试样,若试样水分含量较低(<10%),则取试样直接反复粉碎,至完全过筛。混匀后待测。
若试样水分含量较高(≥10%),试样混匀后,称取适量试样(mC,不少于50 g),置于70±1 ℃真空干燥箱内重复干燥至恒质量。将干燥后试样转至干燥器中,待试样温度降到室温后再称量(mD)。根据干燥前后试样质量,计算试样质量损失因子(f)。干燥后试样反复粉碎至完全过筛,置于干燥器中待测。若试样不宜加热,也可采取冷冻干燥法。
脂肪含量≥10%的试样,需经脱脂处理。称取适量试样(mC,不少于50 g),置于漏斗中,按每克试样25 mL 的比例加入石油醚进行冲洗,连续3 次。脱脂后将试样混匀再按进行干燥称质量记为(mD)。记录脱脂、干燥后试样的质量损失因子(f)。试样反复粉碎至完全过筛,置于干燥器中待测。若试样脂肪含量未知,按先脱脂再干燥粉碎方法处理。
糖含量≥5%的试样,试样需经脱糖处理。称取适量试样(mC,不少于50 g),置于漏斗中,按每克试样10 mL 的比例用85%乙醇溶液冲洗,弃乙醇溶液,连续3 次。脱糖后将试样置于40 ℃烘箱内干燥过夜,称量记为(mD),记录脱糖、干燥后试样的质量损失因子(f)。干样反复粉碎至完全过筛,置于干燥器中待测。
试样总膳食纤维(TDF)测定:在膳食纤维分析仪上安装好试样袋和残渣袋后准确称取双份试样(m)于试样袋中,约1 g(精确至0.1 mg),双份试样质量差≤0.005 g,同时做2 份空白试验。然后在残渣袋(mG)中,各称入硅藻土约1 g(精确至0.1 mg)记为(mDE),根据仪器要求将配置好的试剂放到指定通道后启用仪器,待仪器中间停止后手动调节pH 值,用1 mol/L 氢氧化钠溶液或1 mol/L 盐酸溶液调节试样液pH 值至4.5±0.2。开启仪器继续反应直至仪器运行结束。取下残渣袋分别用78%乙醇15 mL 洗涤残渣袋2 次,用95%乙醇15 mL 洗涤残渣袋2 次,丙酮15 mL 洗涤残渣2 次,沥干去除洗涤液后,将坩埚连同残渣于105 ℃烘干过夜。将坩埚置干燥器中冷却1 h,称量(mGR,包括处理后坩埚质量及残渣质量),精确至0.1 mg。减去处理后坩埚质量,计算试样残渣质量(mR)。蛋白质和灰分的测定,取2 份试样残渣中的1 份按GB 5009.5 测定氮(N)含量,以6.25 为换算系数,计算蛋白质质量(mP);另1 份试样测定灰分,即在525 ℃灰化5 h,于干燥器中冷却,精确称量坩埚总质量(精确至0.1 mg),减去处理后坩埚质量,计算灰分质量(mA)。
1.2 试样中膳食纤维含量的计算

如果试样没有经过干燥、脱脂、脱糖等处理,f=1
总之,互联网+视域下管理会计环节中大数据技术得到了前面的发展,并且大数据技术也推动了管理会计信息数据的多样化、科学护发展,有利于企业展开可持续的经营决策。
式中:mB——试剂空白的质量,g;
mBP——试剂空白残渣中蛋白质的质量,g;
mBA——试剂空白残渣中灰分的质量,g;
mR——试样残渣质量,g;
mDE——硅藻土质量,g;
mGR——恒重残渣袋及残渣质量,g;
mG——恒质量残渣袋质量,g;
m0——恒质量空坩埚质量,g;
mA——试样残渣中灰分的质量,g;
mP——试样残渣中蛋白质的质量,g;
mC——试样制备前的质量,g;
mD——试样制备后的质量,g;
f——试样制备时因干燥、脱脂、脱糖导致质量变化的校正因子;
m——双份试样取样质量值,g;
X——试样中膳食纤维的含量,g/100 g;
V1——试液消耗硫酸或盐酸标准滴定液体积,mL;
V0——试剂空白消耗硫酸或盐酸标准滴定液体积,mL;
C——硫酸或盐酸标准滴定溶液浓度,mol/L。
2 总膳食纤维不确定度来源的关系图[11]
根据膳食纤维计算公式中各参数,不确定度相关的来源分量表示在图1 的关系图中。
总膳食纤维含量不确定度来源的因果关系图见图1。

图1 总膳食纤维含量不确定度来源的因果关系图
3 总膳食纤维不确定度各分量
3.1 总膳食纤维相对标准不确定度urel(f):试样制备时因干燥、脱脂、脱糖导致质量变化的校正因子

3.2 总膳食纤维相对标准不确定度urel(m):双份试样取样质量的均值


3.3 总膳食纤维相对标准不确定度urel(R):双份试样残渣质量的均值
3.4 总膳食纤维相对标准不确定度urel(mA):试样残渣中灰分的质量


3.5 总膳食纤维相对标准不确定度urel(mP):试样残渣中蛋白质的质量
试样残渣中蛋白质的质量[13]带来的不确定度来源为盐酸标准滴定溶液、滴定管。
盐酸标准溶液的浓度为0.100 8 mol/L,标准溶液证书上给出的相对扩展不确定度为0.20%,包含因子k=2,其相对标准不确定度urel(c)=0.20%/2=0.001 000。

4 总膳食纤维不确定度各组分分量汇总[11]
试样制备时因干燥、脱脂、脱糖导致质量变化的校正因子f 带入引起的相对标准不确定度urel(f)、双份试样取样质量均值m¯带入引起的相对标准不确定度urel(m¯)、双份试样残渣质量的均值m¯R带入引起的相对标准不确定度urel(m¯R)、双份试样残渣中灰分的质量mA带入引起的相对标准不确定度urel(mA)、试样残渣中蛋白质的质量mP带入引起的相对标准不确定度urel(mP)、试剂空白的质量mB带入引起的相对标准不确定度urel(mB)。
各测量不确定度分量汇总表见表1。
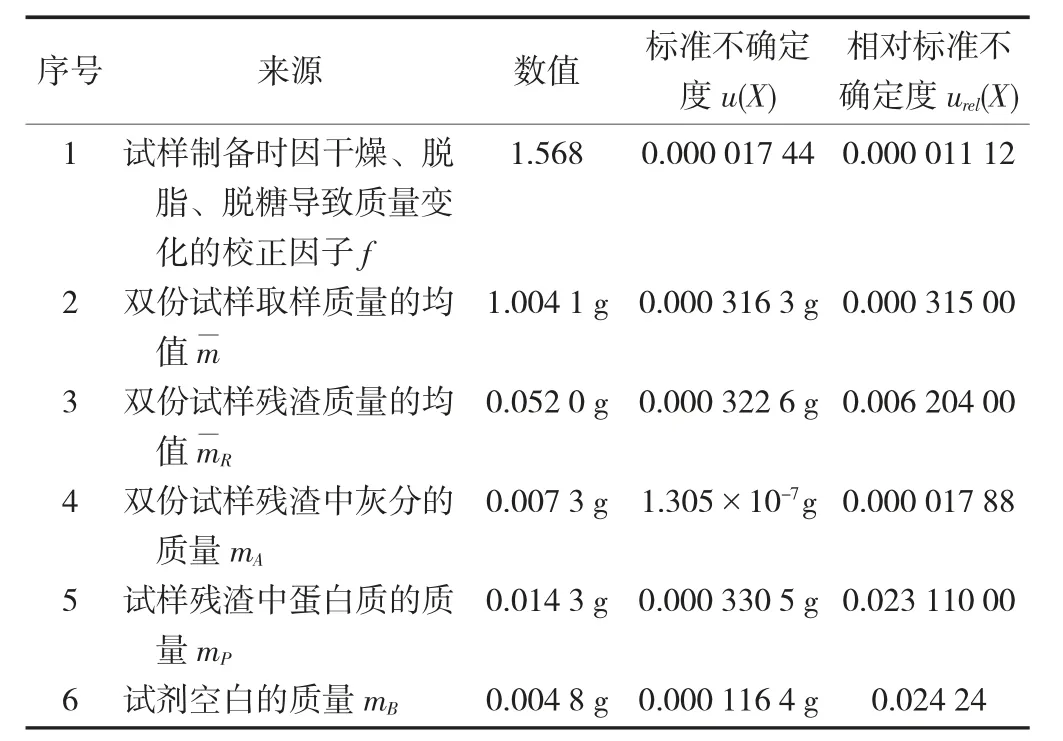
表1 各测量不确定度分量汇总表
5 总膳食纤维合成标准不确定度
依据合成标准不确定度简单规则2 CNAS-GL006:2019:合成总膳食纤维含量相对标准不确定度urel(Hgd)与每个组分有关的测量不确定度,通过计算得到:


6 总膳食纤维扩展不确定度结果完整表示
试样的总膳食纤维含量:

试样的总膳食纤维含量标准不确定度:

总膳食纤维的相对标准不确定度为:
在95%的置信区间,用包含因子k=2 得到的扩展不确定度U95=2×0.078=0.16 g/100 g。
总膳食纤维检测结果的完整表示:
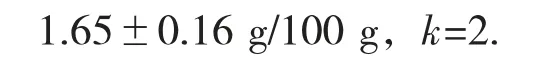
7 结论
试样中总膳食纤维含量可采用手动操作和仪器自动分析来完成。手动操作需对样品进行相对麻烦的前处理,并且所需时间长、各种能耗需求较大、仪器转移损失等缺陷[11]。试验采用全自动膳食纤维仪测定食品中总膳食纤维的含量,日常检测工作操作方便、有效缩短检测周期、定量配制所需试剂、精密度良好,可操作性更优。饼干中总膳食纤维质控样6 次测定结果为1.61~1.68 g/100 g 与证书值1.757±0.438 g/100 g 相符,准确性良好。在满足试验要求的基础上,双份试样质量差≤0.005 g。准确测定灰分和蛋白质的浓度对其定量存在至关重要的作用[14-16]。通过食品中总膳食纤维含量测定结果的影响因素各分量逐个分解,有效减少检测结果相对偏差,使精确度良好,为检测提供一定的参考价值。
猜你喜欢
现代食品(2022年17期)2022-10-20
福建茶叶(2022年8期)2022-10-13
福建交通科技(2022年6期)2022-09-15
林业与环境科学(2022年2期)2022-07-12
发明与创新·大科技(2021年2期)2021-04-20
发明与创新·小学生(2020年6期)2020-06-22
文学港(2016年12期)2017-01-06
健康女性(2016年7期)2016-09-28
故事会(2016年9期)2016-05-06
