
Rethinking the necessity of low glucose intervention for cerebral ischemia/reperfusion injury
2022-11-05 14:29JiahuaXieFarooqahmedKitturAndyLiChiuYuehHung
中国神经再生研究(英文版) 2022年7期
Jiahua Xie, Farooqahmed S. Kittur, P. Andy Li, Chiu-Yueh Hung
Abstract Glucose is the essential and almost exclusive metabolic fuel for the brain. Ischemic stroke caused by a blockage in one or more cerebral arteries quickly leads to a lack of regional cerebral blood supply resulting in severe glucose deprivation with subsequent induction of cellular homeostasis disturbance and eventual neuronal death. To make up ischemiamediated adenosine 5′-triphosphate depletion, glucose in the ischemic penumbra area rapidly enters anaerobic metabolism to produce glycolytic adenosine 5′-triphosphate for cell survival. It appears that an increase in glucose in the ischemic brain would exert favorable effects. This notion is supported by in vitro studies, but generally denied by most in vivo studies. Clinical studies to manage increased blood glucose levels after stroke also failed to show any benefits or even brought out harmful effects while elevated admission blood glucose concentrations frequently correlated with poor outcomes. Surprisingly, strict glycaemic control in clinical practice also failed to yield any beneficial outcome. These controversial results from glucose management studies during the past three decades remain a challenging question of whether glucose intervention is needed for ischemic stroke care. This review provides a brief overview of the roles of cerebral glucose under normal and ischemic conditions and the results of managing glucose levels in non-diabetic patients. Moreover, the relationship between blood glucose and cerebral glucose during the ischemia/reperfusion processes and the potential benefits of low glucose supplements for non-diabetic patients are discussed.
Key Words: blood-brain barrier; blood glucose; cerebral glucose; glucose intervention;glucose transporter; glycosylation; induced hyperglycemia; ischemic penumbra; ischemic stroke; non-diabetic patients
Introduction
Stroke is one of the leading causes of morbidity and mortality worldwide with ischemic stroke accounting for more than 80%of the cases (Feigin et al., 2017). Ischemic stroke is caused by obstruction of cerebral arteries, leading to a lack of regional cerebral blood supply causing severe oxygen and glucose deprivation (OGD), and irreversible neuronal damage and death in the brain tissue (Figure 1) (Robbins and Swanson,2014; Puig et al., 2018). To develop effective therapeutics for ischemic stroke, tremendous efforts have been made to reveal its pathogenic mechanisms and test various treatments(Puig et al., 2018; Huang and Zhang, 2019; Barthels and Das,2020). Currently, mechanical thrombectomy or in combination with thrombolytic agents is major effective treatment, which has significantly improved clinical outcomes. However, no further treatment has been made from bench to bedside since tissue plasminogen activator was approved by the Food and Drug Administration for acute ischemic stroke in 1996.Moreover, less than 5% of stroke patients are eligible for tissue plasminogen activator therapy due to its narrow therapeutic window with multiple contraindications (Barthels and Das,2020) while exploiting neuroprotectants for treatment persists a challenge (Muresanu et al., 2019). Therefore, it is essential to further understand cerebral ischemia-induced pathophysiological changes and develop novel therapeutic strategies for ischemic stroke care.To protect the brain from the ischemia/reperfusion (I/R)injury, various therapeutic strategies, such as inhibiting apoptosis, promoting angiogenesis, suppressing the immune system, repressing reactive oxygen species, and regulating the metabolic processing systems including glucose intervention,have been investigated (Wass and Lanier, 1996; MacDougall and Muir, 2011; Muresanu et al., 2019; Barthels and Das, 2020).Unfortunately, most of these strategies failed in clinical trials while glucose intervention did not show desired effects in either preclinical or clinical studies. There is a possibility that increased glucose in the residual blood flow to the ischemic brain would benefit cell survival because reduced delivery of glucose and oxygen during ischemic stroke causes brain adenosine 5′-triphosphate (ATP) depletion, which affects numerous downstream biological processes resulting in neuronal cell death (Robbins and Swanson, 2014; Barthels and Das, 2020).Although beneficial effects of glucose on neuronal cells have been proved inin vitrostudies three decades ago (Schurr et al., 1987; Callahan et al., 1990), various preclinical studies with animal I/R models and clinical studies with management of elevated blood glucose failed to bring out neuroprotective effects and instead occasionally resulted in adverse effects (Wass and Lanier, 1996; MacDougall and Muir, 2011). In addition,elevated glucose concentrations at admission often correlated with poor outcomes in clinical practice even in patients without prediabetes or diabetes (MacIntyre and Ciechanowski, 2018).Meanwhile, strict glycaemic control in clinical practice also failed to yield any beneficial outcome (MacІntyre and Ciechanowski,2018; Johnston et al., 2019). These controversial results from glucose management studies raise a critical question whether glucose intervention is necessary for ischemic stroke patients.Furthermore, whether hyperglycemia should be corrected and to what extent in patients with acute ischemic stroke remains unanswered.

Figure 1|A schematic diagram of formed ischemic core and ischemic penumbra regions.
This review provides a brief overview of the roles of cerebral glucose under normal and ischemic conditions and the results of glucose management in non-diabetic patients, and finally discusses the relationship between blood glucose and cerebral glucose during the I/R process. With recent progresses in bothin vitroandin vivolow glucose supplement studies and further understanding, the role of glucose in glycosylation, potential benefits of low glucose supplement for non-diabetic patients are discussed. Although prediabetes and diabetes are wellestablished risk factors for stroke, the pathophysiological changes and I/R-mediated injury mechanisms of conditional hyperglycemia-related ischemic stroke are much complex (Venkat et al., 2017). Since as high as 30–40% of initially hospitalized ischemic stroke patients without diabetic conditions were also hyperglycemic (Luitse et al., 2012), in this review, we mainly focus on the roles and metabolism of glucose during ischemia in patients with no pre-diabetic and diabetic conditions.
Search Strategy and Selection Criteria
The studies cited in this review were published from 1987 to 2021, and they were searched on PubMed and Web of Science Databases mainly using the following keywords: stroke,acute ischemic stroke, cerebral ischemia/reperfusion injury,hyperglycemia, hypoglycemia, insulin, cerebral glucose, blood glucose, glucose supplement, glucose intervention, glucose transporter,N-glycosylation,O-glycosylation,O-GlcNAcylation,mitochondrial fission and fusion, and non-diabetic patients.
Brain Has High Energy Demand with Tightly Controlled Glucose Entry
The mammalian brain has a high energy demand (Ritter, 2017;Dienel, 2019). It performs a high aerobic metabolism with the glucose oxidized to CO2and water by consuming six moles of O2for each mole of glucose. Unlike the other organs that can use glucose as well as amino acids and other sources (such as fatty acids, ketone bodies) for energy, the brain mainly uses glucose (Ritter, 2017). The human brain accounts for only ~2%of the body’s weight but consumes ~20% of glucose-derived energy (Ritter, 2017). Therefore, continuous glucose supply is important for neuronal cell survival and brain function.
Both glucose entry from the blood to the brain and its metabolism in the brain under normal physiological conditions are tightly regulated (Mergenthaler et al., 2013; Zhang et al., 2014; Koepsell, 2020). The blood-brain barrier (BBB) and glucose transporters (GLUTs) play critical roles in controlling glucose transport to the brain (Figure 2). The BBB selectively controls inward and outward transportation of glucose,oxygen, and many other substances through cell junctions and cell surface transport systems and several GLUTs. Among 15 identified GLUTs, GLUT1 and GLUT3 are considered to be the primary transporters for glucose uptake in the brain(Koepsell, 2020). GLUT1 expression form with a molecular weight of 55-kDa has been confirmed to be highly enriched in the microvascular endothelial cells of the BBB while GLUT3 is predominantly expressed in neurons with a higher transport rate to facilitate neuronal glucose uptake (Zhang et al., 2014;Koepsell, 2020). In addition to glucose transport, some cerebral glucose transporters also play a critical role in sensing glucose concentrations in the blood and brain. GLUT2 and GLUT4 are expressed in neurons and astrocytes where they also act as glucose sensors and are involved in the regulation of glucose homeostasis. Furthermore, GLUT5, GLUT6, and GLUT8 were found to be expressed in various regions of the brain, but the physiological roles of these as well as the remaining other members in the brain are still unclear (Zhang et al. 2014; Koepsell, 2020).
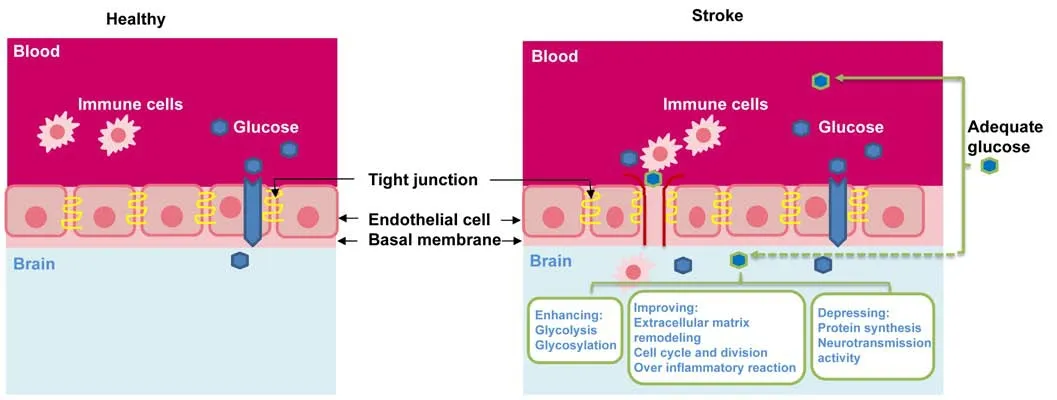
Figure 2|Possible benefits from low glucose supply via blood glucose management or onsite low glucose supply.
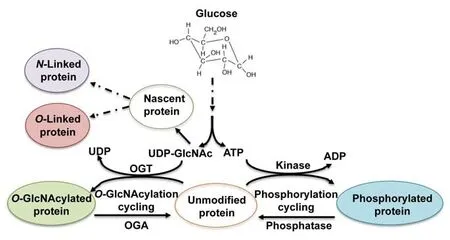
Figure 3|Utilization of UDP-GlcNAc synthesized from glucose in protein glycosylation.
In general, cerebral glucose levels are maintained much lower than blood glucose levels (Meierhans et al., 2010; Ritter,2017). During euglycemic conditions (about 5 mM glucose peripherally), the brain glucose concentrations range between 1 to 2.5 mM and may vary depending on different regions of the brain (Robbins and Swanson, 2014; Ritter, 2017). Once the blood glucose levels are elevated, the cerebral glucose intake is significantly decreased in order to maintain the normal cerebral metabolic levels (Meierhans et al., 2010). Therefore,the integrity of BBB and the proper expression and function of GLUTs are crucial for the regulation of cerebral glucose homeostasis.
Functions of Metabolites Derived from Cerebral Glucose Metabolism
Continuous supply of glucose from the circulation system to the brain is critical to fulfill many essential functions, including ATP generation, oxidative stress management, and synthesis of neurotransmitters, neuromodulators, and structural components (Dienel, 2019). Glucose metabolism mainly generates ATP to provide energy for brain functions with the largest proportion of produced energy consumed for synaptic activity, such as, generating action and postsynaptic potentials,and maintaining ion gradients and neuronal resting potential(Mergenthaler et al., 2013). Additionally, glucose metabolism also provides precursors and ATP for the biosynthesis of neurotransmitters (Dienel, 2019) since neuroactive compounds(e.g., glutamate, aspartate, glycine, D-serine) are blocked strictly by the BBB and must be synthesized from glucose within the brain (Mergenthaler et al., 2013). Glucose is also required for oxidative stress management through both quenching and the production of reactive oxygen species in the central nervous system (Magistretti and Allaman, 2018; Dienel,2019). Glucose metabolism generates nicotinamide adenine dinucleotide phosphate through the pentose phosphate pathway (Magistretti and Allaman, 2018). The nicotinamide adenine dinucleotide phosphate produced from this pathway is used by glutathione reductase to regenerate glutathione from glutathione disulfide for dealing with oxidative stress whereas it can also be used by nitric oxide synthase to produce nitric oxide (NO˙) or by nicotinamide adenine dinucleotide phosphate oxidase to produce superoxide (O2˙) (Robbins and Swanson, 2014; Dienel, 2019). Furthermore, brain glucose metabolism is also linked to cell death pathways via glucosemetabolizing enzymes. For example, hexokinase II, a hypoxia up-regulated hexokinase isoform in the brain, has been demonstrated to inhibit apoptosis in the presence of available glucose, but promote neuronal cell death under glucose deprivation (Mergenthaler et al., 2013). Thus, disruption of glucose delivery and metabolism leads to debilitating brain diseases, affecting nerve cell survival, and impairing brain function.
Cerebral Glucose Is Essential for Protein Glycosylation
Cerebral glucose also plays important regulatory roles in glycosylation (Figure 3) (Videira et al., 2018; Paprocka et al.,2021).Glycosylation, includingN-glycosylation,O-glycosylation,andO-GlcNAcylation, is an important post-translational modification that determines protein folding, solubility,secretion, circulatory half-life, and protein functionality while nearly all membrane proteins are glycoproteins (Dennis et al., 2009; Bukke et al., 2020).N-glycosylation is a process of the attachment of a complex glycan chain (oligosaccharide)to nitrogen of asparagine residue whileO-glycosylation involves labeling of the hydroxyl group of mainly serine(Ser) or threonine (Thr) residues with the glycan chain.Glucose is required forde novouridine diphosphate-Nacetylglucosamine (UDP-GlcNAc) biosynthesis by hexosamine biosynthetic pathway (HBP), which is a precursor to cytidine monophosphate-sialic acid, the sugar-nucleotide donor utilized by sialyltransferases to capN- andO-glycans (Dennis et al., 2009). Proper proteinN-glycosylation is essential to sustain neuronal migration, axon guidance, and synaptic physiology,which play vital roles in brain development (Paprocka et al.2021).O-glycosylation comprises of different classes involving attachment of several modifying sugars (Fuc, Glc, GalNAc,GlcNAc, Man, or Xyl) linked to hydroxyl group of Ser, Thr or Tyr, and is involved in diverse biological processes. Compared toN-glycosylation, current knowledge ofO-glycosylation is still limited because of its diversity and complexity, and the lack of effective analysis methods (Darula and Medzihradszky,2018). The major biological roles ofO-glycosylation have not been elucidated yet, but it was found to function in proteinprotein interactions, intercellular signaling and protein folding,interaction with pathogens, cell adhesion, and proteolytic processing (Darula and Medzihradszky, 2018). Previous studies also revealed thatO-glycosylation is necessary for many brain proteins (Trinidad et al., 2013).
In contrast to the complexN- andO-linked glycosylation that requires several enzymes to modify target proteins irreversibly with glycan chains,O-GlcNAcylation is a non-canonicalO-glycosylation whereinO-linkedN-acetylglucosamine(O-GlcNAc) moieties are reversibly attached to Ser and Thr residues of cytoplasmic, nuclear and mitochondrial proteins.This distinct glycosylation is regulated by two enzymes:O-GlcNAc transferase andO-GlcNAcase.O-GlcNAc transferase catalyzes the transfer of anO-GlcNAc moiety from the donor substrate UDP-GlcNAc to the hydroxyl groups of target Ser and Thr residues, whereasO-GlcNAcase removes theO-GlcNAc from the proteins (Figure 3) (Hart et al., 2007).O-GlcNAcylation is a highly dynamic and rapidly reversible in response to various environmental stresses (Hart et al., 2007;Thompson et al., 2018). In this regard,O-GlcNAcylation is an essential post-translational modification, which interplays with other post-translational modifications including phosphorylation in a reciprocal manner to compete with phosphorylation sites (van der Laarse et al., 2018). UDPGlcNAc biosynthesized from glucose via the HBP is a key substrate for proteinO-GlcNAcylation (Hart et al., 2007). Іt is not difficult to understand thatO-GlcNAc is closely regulated by cellular glucose concentrations since 2 to 3% of total glucose is transformed into UDP-GlcNAc (Ryan et al., 2019). In addition, the protein phosphorylation requires ATP produced from glycolysis and tricarboxylic acid cycle, and glucose levels regulate protein phosphorylation andO-GlcNAcylation through ATP to affect the activity of kinases and phosphatases(Gatta et al., 2016; Ryan et al., 2019).
So far, more than 4500O-GlcNAc modified proteins have been identified and known to involve in all aspects of cellular metabolism (Hart et al., 2007; Ma et al., 2020).Among them, there are thousands ofO-GlcNAc-modified proteins identified in the mammalian brain (Thompson et al.,2018). Accumulated evidence shows thatO-GlcNAcylation not only plays important physiological roles in neuronal health and function but also displays critical roles in several neurodegeneration diseases through the involvement of glucose sensing, signal transduction, the stress response,cell division as well as regulating gene expression, protein stability, metabolism, mitochondrial fission and fusion, and so on (Rexach et al. 2008; Hwang and Rhim, 2018; Thompson et al., 2018). Maintaining the balance of mitochondrial fission and fusion is especially important for neurons because they require continuous redistribution of mitochondria across long distance to meet end-to-end energy requirements, Ca2+buffering, and neurotransmission (Balog et al., 2016; Liu et al.,2018; Bukke et al., 2020; Sabouny and Shutt, 2020). Besides,O-GlcNAc transferase cytosolic activity is ten times higher in the brain when compared to other tissues (Okuyama and Marshall, 2003). Therefore,O-GlcNAcylation with its activity tightly linked to glucose content is essential for neuron survival, maintenance, and function (Wang et al., 2016). In sum, maintaining glucose levels is important and reduced cerebral glucose levels may affect glycosylation and impair glycoprotein functions, which in turn damage nerve cells and compromise brain function.
Cerebral Glucose during Ischemia
Anaerobic metabolism of glucose in the ischemic penumbra region
When ischemia occurs, regional cerebral blood flow (CBF)levels fall, which form three zones in the ischemic brain:ischemic core, ischemic penumbra, and non-ischemic regions based on the CBF gradient (Figure 1). Cells within the ischemic core where the CBF declines below the threshold of energy failure (15–20%), are irreversibly damaged within a few minutes after stroke (Ahnstedt et al., 2016). They are unrescuable and undergo necrotic cell death accompanying the release of glutamate into the neighboring ischemic penumbra region where it induces their excitotoxic damage(Pisani et al., 2004). On the contrary, the ischemic penumbra is an area around the ischemic core where cells have impaired functions with the absence of electrical potentials but are metabolically viable and potentially salvageable within a certain time window if local blood flow is reestablished (Heiss,2011). However, the penumbra progressively experiences irreversible damage and enters into a necrotic core unless reperfusion treatment is applied timely (Heiss, 2011). For this reason, the ischemic penumbra is the main target of acute therapeutic interventions for the treatment of ischemic stroke.
In the ischemic penumbra region, impaired CBF leads to severe OGD. Since the brain’s demand for energy is high and neurons lack glycogen stores, glucose in the ischemic penumbra area supplied by the residual blood flow enters anaerobic metabolism rapidly to produce glycolytic ATP for cell survival even in the absence of oxygen (Robbins and Swanson, 2014). However, this anaerobic glucose metabolism also produces lactic acid, which together with glucose-fueled ATP hydrolysis during reperfusion to release H+coupled with a lack of mitochondrial H+consumption contributes to reduced pH of brain tissues and exacerbates brain injury through impaired antioxidant enzyme functions (Suh et al., 2008). Within minutes after ischemia, OGD-induced mitochondrial dysfunction also occurs, resulting in depletion of ATP production and overproduction of reactive oxygen species (Mergenthaler et al., 2013). This ATP depletion is one of the major initiators to trigger dysregulation of numerous processes, such as membrane ion pump failure to efflux of cellular potassium, influx of sodium, chloride, and water, and membrane depolarization (Mergenthaler et al., 2013; Robbins and Swanson, 2014), resulting in neuronal cell damage and loss. In light of this, it would seem to follow that increased cerebral glucose levels to subsequently enhance glucose content in the ischemic penumbra could improve cell survival by providing glycolytic ATP. However, glucose management in animal models and clinical studies has neither improved functional outcomes nor reduced mortality, or even cause worse outcomes (detailed see following sections). These results imply that it is a challenge to understand the roles of glucose in ischemic stroke.
Hyperglycemia in ischemic stroke
It has been observed that the vast majority of cerebral ischemic stroke patients have abnormal regulation of glucose metabolism (Matz et al., 2006). Blood glucose is usually increased in the hours after a stroke (Gray et al., 2007; Everettand Mathioudakis, 2018). In both humans and rodents, the typical blood glucose levels are ~5 mM (Robbins and Swanson,2014; Ritter, 2017). Considering blood glucose levels > 6.1 mM as hyperglycemic, 30–40% of the non-diabetic ischemic stroke patients are hyperglycemia at admission (Luitse et al.,2012). This hyperglycemic phenomenon has been considered as the result of a stress response to the stroke event (Gray et al., 2007; Everett and Mathioudakis, 2018). Іt was noticed that hyperglycemia at admission was associated with poor outcomes in stroke patients undergoing thrombolytic therapy(Everett and Mathioudakis, 2018). The undesirable outcomes associated with hyperglycemia could possibly result from the acidification of affected brain areas, enhancement of glucosesodium exchange, amplification of inflammatory response,and formation of abnormal protein glycosylation as well as glucose-induced overproduction of reactive oxygen species and activation of endothelial protein kinase C (Robbins and Swanson, 2014). All these changes could disturb recanalization and increase reperfusion injury leading to further ischemic damage or causing hemorrhagic transformation of the infarct region.
Previous studies have suggested that hyperglycemia promotes ischemic injury by various mechanisms, including increased mitochondrial fragmentation, amplified oxidative stress by glucose-fueled superoxide production, impaired antioxidative capacity, enhanced glucose-sodium exchange and formation of abnormal protein glycosylation, and advanced glycation products (Suh et al., 2008; Anzell et al., 2018; Jensen et al., 2019; Yang et al., 2019; Sabouny and Shutt, 2020).Mitochondria are both a source and target of I/R injury and cell death, and that the imbalance of mitochondrial fission and fusion is one of the hallmarks of I/R induced neuronal death by impairing mitochondria functions (Balog et al.,2016; Anzell et al., 2018; Liu et al., 2018). Hyperglycemia is known to promote mitochondrial fragmentation to disturb mitochondrial dynamic balance (Liesa and Shirihai, 2013).This hyperglycemia-mediated mitochondrial fragmentation is believed to occur viaO-GlcNAcylation modifications of key mitochondrial fission-related protein dynamin-related protein 1 and fusion-related protein optic atrophy protein 1 under transiently elevated high sugar levels (Hart et al., 2007). In neonatal cardiomyocytes, high glucose has been reported to promote dynamin-related protein 1O-GlcNAcylation and increase its stability resulting in enhanced mitochondrial fission while optic atrophy protein 1O-GlcNAcylation leads to its breakdown causing impaired mitochondrial fusion (Klimova et al., 2018). Under I/R-induced hyperglycemic conditions,glutathione production is also impaired, which in turn compromises the capacity of the brain to counter oxidative stress (Robbins and Swanson, 2014).
All these above studies suggest that cerebral I/R injuryinduced hyperglycemia should be intervened. However, strict glycaemic control has failed to yield any beneficial outcome(Everett and Mathioudakis, 2018; Johnston et al., 2019).Therefore, further understanding of pathophysiological mechanisms of cerebral ischemic stroke-mediated hyperglycemia is necessary. Alternatively, the observed high percentage of non-diabetic stroke patients exhibiting hyperglycemia might be the result of body’s response to compensate for lower glucose in ischemic tissues in order to promote cell survival as recently suggested by Li et al. (2020).
Glucose Management by Either Increasing or Lowering Levels Has Failed to Display Any Neuroprotective Effects
As discussed above, reduction of CBF during ischemia restricts the delivery of oxygen and glucose, and the glucose from the residual blood flow in the ischemic penumbra region is consumed through anaerobic metabolism to produce ATP and some other essential compounds for rescuing cells within this region (Robbins and Swanson, 2014; Dienel, 2019).This implies that supplying additional glucose would benefit neuronal cell survival and function during ischemia, which were reinforced byin vitroprotective studies of glucose on neuronal cells three decades ago (Schurr et al., 1987; Callahan et al., 1990) and our own recent study (Li et al., 2020).However, the results of thesein vitrostudies are contradictory with those fromin vivoand clinical studies. Studies using various І/R animal models indicated that increasing circulating blood glucose by intravenous infusion exacerbated brain injury and mortality compared with normoglycemic control(MacDougall and Muir, 2011; Robbins and Swanson, 2014).The same is true in clinical trials when the intravenous infusion to increase circulating blood glucose failed to present any beneficial effects or even displayed harmful effects (MacDougall and Muir, 2011). Increasing evidences have also shown that hyperglycemia has been identified as a predictor of poor outcome (Luitse et al., 2012; Everett and Mathioudakis, 2018; MacIntyre and Ciechanowski, 2018). All thesein vivostudies indicate that increasing blood glucose is not a good idea for ischemic stroke care. For a number of years, tight glycemic control has been recommended at least for critically ill patients (Everett and Mathioudakis, 2018).
To achieve normoglycaemia in the early stage of stroke, the glucose-potassium-insulin infusion in the first 24 hours of stroke onset was applied to maintain blood glucose levels within the range of 70–130 mg/dL (3.89–7.21 mM) (Gray et al.,2007; Bellolio et al., 2012). Unfortunately, no positive effects were observed by lowering blood glucose levels. Conversely,the risk of inducing potentially harmful hypoglycemia was high in patients subjected to tight glucose control regimens(MacІntyre and Ciechanowski, 2018). Іn addition, two metaanalyses with seven and 22 eligible randomized clinical trials,respectively, showed that using insulin to intensively control blood glucose level in the first hour of stroke did not improve either the functional outcome or the mortality, and instead significantly increased the risk of hypoglycemia (Bellolio et al., 2012; Ling et al., 2012). Nevertheless, the failure of both therapeutic attempts at increasing or lowering glucose left a controversial view about glucose intervention for patients with ischemic stroke. Although the high frequency of hyperglycemia was observed at the admission of non-diabetic patients (Luitse et al., 2012), it remains uncertain whether hyperglycemia should be corrected.
Rethinking the Necessity of Glucose Supplement in Stroke Management
Blood and cerebral glucose levels
To answer whether glucose intervention is necessary or not,it would be helpful to understand the relationship between blood and cerebral glucose levels. Currently, most animal and clinical studies on the beneficial effects of glucose and correcting hyperglycemia in ischemic stroke have focused on blood glucose changes while very less attention has been paid to cerebral glucose levels (Robbins and Swanson, 2014;Zhang et al., 2014). Glucose transport to the brain is tightly controlled by GLUTs. Therefore, the cerebral glucose content plays a more critical and direct role in the ischemic penumbra region than blood glucose level.
When considering cerebral glucose content and its metabolism, an additional glucose supply could benefit cell survival in the ischemic penumbra region. Under normal physiological conditions, the cerebral glucose concentration is only 20% to 50% of that in the blood (Ritter, 2017). Once brain CBF levels fall after ischemic stroke, the cerebral glucose contents drop abruptly, which might result in lower extracellular glucose levels in the ischemic penumbra than in the non-ischemic region. Theoretically, increased glucose supply would compensate for ischemia-induced reduction in extracellular glucose levels in the ischemic penumbra zone to promote cell survival. This is well supported byin vitrostudies showing that different levels of glucose supply under hypoxic conditions could significantly reduce hypoxia-induced neuronal cell damage and death through various mechanisms compared to OGD treatment (Schurr et al., 1987; Callahan et al., 1990; Li et al., 2020). Under hypoxic conditions, neurons like cancer cells depend exclusively on glucose glycolytic metabolism for energy generation to prevent apoptosis(Mergenthaler et al., 2013). In both cell types, hexokinase II plays an important role in regulating apoptosis by switching its apoptotic effect under glucose deprivation to anti-apoptotic effect with available glucose via increasing the activity of the pentose phosphate pathway to prevent oxidative stressmediated cellular damage and inhibit cytochrome c-mediated apoptosis (Mergenthaler et al., 2013).
Additionally, around one-third of non-diabetic stroke patients were hyperglycemic at admission (Luitse et al., 2012; Zhang et al., 2014). This phenomenon could also imply that quickly elevating blood glucose levels by ischemia is one of the possible acute response mechanisms to compensate for low glucose in ischemic tissues during the onset of ischemia.Both GLUT1 and GLUT3 were observed to be induced at transcriptional and translational levels after 1.5 hours of initiating ischemia and lasting up to 24 hours (Espinoza-Rojo et al., 2010). Under cerebral ischemia, GLUT3 protein is mainly expressed in penumbral regions (Ginsberg, 1990). These augmented GLUTs support that enhanced glucose transport activity is needed during this early stage. Transiently induced hyperglycemia could be related to protective mechanisms to enhance glucose transport to the brain.
Yet, glucose intervention on ischemic stroke patients by elevated blood glucose concentrations produced detrimental side effects (MacDougall and Muir, 2011). The pathophysiological mechanisms for this failure have not been clearly clarified, but І/R-mediated disruption of the BBB and elevated blood glucose levels could be mainly responsible. The BBB is a diffusion barrier that allows hydrophobic molecules and metabolic products to be passed by passive diffusion,but prevents the entry of microscopic substances, hydrophilic molecules, and potential neurotoxins (Jayaraj et al., 2019).Between the cerebral endothelial cells, a diffusion barrier is formed by tight junctions to block blood-borne substances to enter the brain. During ischemia, the BBB is disrupted(Figure 2) and loses its barrier capacity. A damaged BBB then is permeable to some large molecules, such as inflammatory factors, neurotoxins, and pathogens, into the brain (Jayaraj et al., 2019). There is accumulating evidence indicating that a ruptured BBB allows peripheral adaptive and innate immune cells, including monocytes, neutrophils, and different types of T cells and B cells to enter the central nervous system(CNS), where they perform distinctly different type cellmediated either neuroprotective, neurotoxic or both effects(Bonaventura et al., 2016; Prinz and Priller, 2017). Reperfusion to re-establish blood flow also worsens the barrier function and induces more endothelium damage, microcirculation obstruction, and immune cell infiltration, and complement system activation to propagate inflammation (Bonaventura et al., 2016). Transferring of blood-borne immune cells through the BBB into the CNS during ischemia has been considered a key process in neuroinflammation (Bonaventura et al., 2016).Following ischemic stroke, microglia and astrocytes were found to be activated within hours leading to the production of cytokines and chemokines while the infiltration of immune cells into the brain is induced by both I/R injuries (Jayaraj et al., 2019). It was observed that elevated circulating glucose concentrations could worsen the barrier function (Huang et al., 1996). Hence, increasing blood glucose at an adequate level (remaining undetermined at this time) may facilitate CBF to exert beneficial effects, but improperly elevating blood glucose may cause more BBB damage and allow more of the restricted blood-borne substances and cells to pass through and magnify ischemia-induced injury.
Beneficial effects of low levels of glucose supplement
In clinical practice, only persistent hyperglycemia (lasting> 24 hours after stroke) was highly associated with poorer outcomes whereas this was not seen in patients who had transient hyperglycemia on the initial examination (Zhang et al., 2014; Everett and Mathioudakis, 2018). This observation may suggest that the unfavorable influence of hyperglycemia on the outcome is only linked to the patients who are hyperglycemic already at stroke onset rather than those who experience transient hyperglycemia responding to a stress in the hours after the stroke. To further determine detrimental or beneficial effects of transiently induced hyperglycemia and observe corresponding cerebral glucose level changes, Zhang et al. (2016) used an I/R rat model to transiently increase blood glucose levels by intraperitoneal injection with different doses of glucose. When a single dose of 0.5, 1, 1.5, or 2 g/kg glucose was injected before reperfusion after 90 minutes of ischemia, blood glucose levels were transiently elevated by all these treatments to reach the peak levels of 6 to 16 mM after reperfusion for 30 minutes, and then returned to the level of no glucose control after reperfusion for 2 hours. Compared to the sham, I/R alone (without glucose) or I/R with 2 g/kg glucose treatment caused significant-high glucose contents in both cortex and striatum after reperfusion for 24 hours. Іn the striatum, glucose contents in these two treatments were still maintained at significantly high levels after reperfusion for 28 days. However, 1 g/kg glucose treatment restored I/R-induced high glucose contents in cortex and striatum to sham levels after reperfusion for 24 hours, maintained better BBB integrity,and displayed excellent short- and long-term neuroprotective effects with high survival rates compared to no glucose control treatment whereas 2 g/kg glucose treatment cause harmful effects (Zhang et al., 2016). The studies indicated that intervention with glucose in certain concentration window has neuroprotective effects even though the hyperglycemia was observed during initial ischemia.
Very recently, ourin vitrostudy showed that 0.69 mM low glucose supplement (hyclone DMEM/high medium glucose level 22 mM) is protective to neuronal cells compared to no glucose control under hypoxic conditions, and that this low glucose treatment promotes cell survival by multiple mechanisms (Figure 2) (Li et al., 2020). Under hypoxic conditions, low-level glucose supply activated hypoxiainducible factor 1 subunit alpha to induce a group of its target proteins and enhance glycolysis to benefit cell survival through further inducing the expression levels of glycolytic pathway genes whose expressions have been already induced by OGD. This low glucose supply could also prevent the dysregulations of extracellular matrix remodeling, cell cycle and division, and antioxidant and detoxification. In addition,the treatment could adjust the over-response of inflammatory reaction proteins to OGD stress, and suppress protein synthesis and neurotransmission activity to promote cell survival. Іn our study, all differentially abundant proteins in the carbohydrate binding category were abundance-increased,which are involved in glycolysis, andN- andO-glycosylation,suggesting that cells under low glucose supply can enhance glucose metabolism and/or protein glycosylation to adapt to OGD stress. The finding from this study could facilitate understanding how low glucose supply affects different cellular pathways in OGD stressed neuronal cells and designing new strategies to promote neuroprotection.
Challenges for Future Research
From this brief overview of glucose metabolism and its roles under normal and ischemic conditions, significant progress has been made in the area while the major question regarding necessity of glucose intervention for ischemic stroke patients remains unanswered. Based on the above discussion,the demonstrated neuroprotective effects of 1 g/kg glucose treatment but harmful effects of 2 g/kg treatment(Zhang et al., 2016) warrant more studies in other I/R animal models and further optimization of glucose concentration and administration time. Asin vitrolow glucose supply can regulate multiple cellular pathways to benefit cell survival (Li et al., 2020), performing genome-wide studies on the penumbral tissues from 1 g/kg glucose treatment at both transcriptional and translational levels could reveal neuroprotective mechanisms in more detail. Transcriptomic and proteomic results, including glycosylation modification,can be used to understand how certain levels of glucose treatment could benefit neuronal cells and further extend these studies to human subjects.
In addition, GLUT family members play important roles in brain for glucose transport and sensing. Only GLUT1 and GLUT3 have been characterized as the primary transporters for glucose uptake in the brain (Koepsell, 2020). Since several members have been found to be expressed in different areas of the brain, further analysis of their specific expression patterns and sites during ischemia along with glucose treatment will facilitate our understanding of their roles in glucose utilization, relations with transiently induced hyperglycemia, and any beneficial effects of low glucose treatment.
Author contributions:JX conceived the manuscript. All authors reviewed the literature, wrote the manuscript, and approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:This work was supported by a grant from the National Institute of General Medical Sciences (SC1GM111178) to JX.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Microglia activation, classification and microgliamediated neuroinflammatory modulators in subarachnoid hemorrhage
- MicroRNA biomarkers in frontotemporal dementia and to distinguish from Alzheimer’s disease and amyotrophic lateral sclerosis
- Protein synthesis modulation as a therapeutic approach for amyotrophic lateral sclerosis and frontotemporal dementia
- A novel viewpoint in glaucoma therapeutics: enriched environment
- Neuronal reprogramming in treating spinal cord injury
- The role of L-arginine metabolism in neurocritical care patients