
纤维素及其衍生物催化脱氧转化制化学品
2022-10-19 10:09:44王伟王瑶占自祥谭天邓卫平张庆红王野
物理化学学报 2022年10期
王伟,王瑶,占自祥,谭天,邓卫平,张庆红,王野
厦门大学化学化工学院,醇醚酯化工清洁生产国家工程实验室,能源材料化学协同创新中心,固体表面物理化学国家重点实验室,福建 厦门 361005
1 Introduction
Sustainable and efficient synthesis of chemicals and fuels from renewable biomass represents an attractive strategy to reduce our reliance on fossil resources (e.g., petroleum, coal and natural gas)1-10. Particularly, catalytic transformation of lignocellulosic biomass, the most abundant biomass in nature,into value-added chemicals or fuels can meet the requirements of green chemistry and help to build sustainable chemical industries11. Theoretically, lignocellulosic biomass has the potential to substitute for fossil resources to synthesize a broad range of chemicals. However, over-oxygenated functional groups with high oxygen contents (~40%, mass fraction) exist in lignocellulosic biomass12, making the biomass conversion significantly different from the current petroleum refineries. To obtain basic chemicals, it generally requires the selective oxidation or other functionalization process in the petrochemical industry, whereas in the conversion of biomass, excess oxygen atoms in biomass should be selectively removed by deoxygenation. Thus, the efficient catalytic systems in conventional petroleum refinery cannot be directly applied to the biomass valorization, and the development of distinctive catalysts and deoxygenation strategies for biomass transformations are urgently desirable. Although gasification and pyrolysis or liquefication can convert biomass to syngas(CO/H2mixture) and bio-oils, further transformation of syngas via Fischer-Tropsch synthesis or upgrading of the bio-oils by hydrodeoxygenation is needed to offer bulk chemicals or hydrocarbon fuels. Compared to these thermochemical processes, selective transformation of biomass and its derived platform compounds under mild conditions may avoid high energy consumption and low efficiency, and is more appealing,since the high selectivity towards target products is achievable via catalysis.
Cellulose is a polysaccharide of glucose connected via the stable β-1,4-glycosidic bonds, and accounts for 35%-50% weight percent in lignocellulosic biomass. Therefore, efficient utilization of cellulose holds a key to biomass valorization. Due to the presence of β-1,4-glucosidic linkage, cellulose molecules arrange in a chain-like manner, which facilitates the interaction between each polymer chain. Moreover, large numbers of hydroxyl groups inside cellulose lead to the formation of extensive intra- and inter-molecular hydrogen bonding between the glycosidic linkages and hydroxyl groups, restricting the accessibility of chemical bonds by the active sites in catalysts,particularly for heterogeneous catalysts.
A variety of acid-based catalysts have been developed in the past decades for the hydrolysis of cellulose13-15, and achieved high efficiency in the formation of glucose, which makes it feasible for the further conversion of cellulose via glucose platform. Given the presence of over-functionalized oxygencontaining groups (i.e., a carbonyl and five hydroxyl groups)inside glucose, it is essential to deoxygenate specific functional groups to produce value-added chemicals from cellulose and/or glucose. For example, the synthesis of ethanol requires the conversion of cellulose/glucose into C2intermediates and selectively cleaving one C-OH bond via hydrogenolysis16-20(Fig. 1). The removal of two or more hydroxyl groups inside glucose and glucose-derived sugar acids (e.g., glucaric acid) will generate oxygen-reduced sugars and even industrial monomers(e.g., adipic acid) (Fig. 1). Besides glucose, HMF that is obtainable from the cascade transformation of cellulose via hydrolysis and subsequent dehydration21, serves as another important bio-platform to synthesize chemicals. Since HMF possesses several different oxygen-containing groups, precisely activating and removing particular oxygen atoms from HMF can provide different valuable chemicals. For example, 2,5-dimethyl furfural (DMF) can be produced by the hydrogenolysis of hydroxyl and carbonyl groups that link furan ring. If the furan ring is opened through cleaving the C-O-C ether bonds, 1,6-hexanediol (1,6-HD), a monomer product, would be formed(Fig. 1).
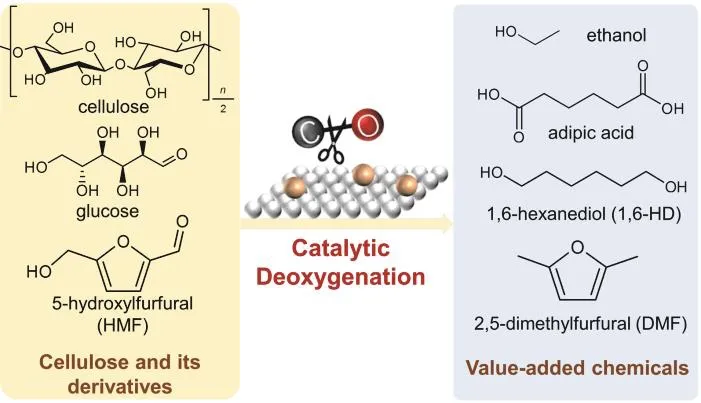
Fig. 1 Catalytic deoxygenation of cellulose and its derivatives into value-added chemicals.
In the past decades, an array of heterogeneous catalysts has been developed and applied to the catalytic deoxygenation of cellulose and its derivatives, achieving precise control over the selective scission of C-O bonds. Although a large number of chemicals can be obtained from cellulose and its derivatives,herein, we only overview recent progress in the selective transformation of cellulose, glucose, HMF, and their derived compounds into ethanol, DMF, 1,6-HD, and adipic acid, with an emphasis on the heterogeneous catalysis. It is not only because these chemicals are of high values, but also due to the fact that the formation of these chemicals represents the cleavage of different C-O bonds in biomass molecules. The active sites of typical catalysts and reaction mechanisms have been presented to get insight into the C-O bond cleavage in these biomass molecules. In addition, other factors such as reaction conditions that affect the deoxygenation performance are analyzed as well.We hope that these knowledge gained on the catalytic deoxygenation of cellulose and its derived platforms will promote the rational design of effective strategies or catalysts in the future utilization of lignocellulosic biomass.
2 Hydrogenolysis of cellulose to ethanol via selectively cleaving a C-OH bond
Ethanol, a simple C2alcohol, has been used as a versatile building block to produce many valuable chemicals, and has also been regarded as an alternative to gasoline22. Currently, ethanol is mainly produced from the fermentation of edible carbohydrates such as glucose or starch. Pyruvate, a C3sugar, is formed in this fermentation process and acts as a key intermediate, which undergoes further decarboxylation to give ethanol and equivalent amount of CO2(Fig. 2a)23, Because CO2was generated during this fermentation, the maximum ethanol yield is limited to 67% on the basis of carbon. In contrast,chemocatalytic approach may avoid CO2generation through tuning the reaction pathways, and hence have potential to achieve high atom efficiency.

Fig. 2 Three typical reaction methods for the synthesis of ethanol from cellulose 16. (a) Biological process; (b) Two-step route via methyl glycolate intermediate; (c) One-pot conversion of cellulose to ethanol.
A typical example for the chemocatalytic method is the conversion of cellulose to ethanol through a two-step route(Fig. 2b)24,25. Initially, cellulose undergoes an oxidative C-C bond scission to form methyl glycolate (MG) in methanol solvent. Upon hydrogenation or hydrogenolysis of one C-OH bond, the formed MG is subsequently converted to ethanol.Tungsten oxide was reported as an active catalyst in the first step,giving a MG yield of ~60% under the conditions of 1 MPa O2and 513 K24, while Pt-modified Cu/SiO2catalyst was capable of catalyzing the deoxygenation of MG to ethanol with a yield of 77% at 503 K in 3 MPa H225. A total ethanol yield from the twostep route could reach ~46%, which was comparable to the fermentation process. With improving the catalytic performances of both catalysts in this two-step route, the ethanol yield can be further enhanced, which may make the process more competitive than the mature fermentation process. However,energy-consuming separation of MG from the first oxidation reaction is a necessary step, which consequently makes the whole process costly. Moreover, such chemical route employs methanol as reaction medium in the oxidation step, in which the methanol oxidation might occur, resulting in resources waste and causing potential explosion risk in large-scale application.
As an alternative to the two-step route, one-pot conversion of cellulose to ethanol is more promising. In this approach,cellulose undergoes consecutive hydrolysis and retro-aldol reaction to afford C2compounds (e.g., glycolaldehyde, ethylene glycol), and further hydrogenolysis of these C2intermediates can generate ethanol (Fig. 2c). Since the selectively cleaving both C-C and C-OH bonds is involved in this process, the development of multifunctional catalysts holds the key.Tungsten-based compounds (e.g., H2WO4) have been widely utilized in cutting the C-C bond inside cellulose for the production of C2products such as ethylene glycol. Hence,combining a tungsten-based compound with an active metal that enables hydrogenolysis of C-OH bonds will be expected to succeed in catalyzing the one-pot transformation of cellulose into ethanol. For instance, Wang and co-workers simply combined H2WO4and Pt/ZrO2, and successfully synthesized ethanol from direct reaction of cellulose in H2O under H2(Fig. 2c)16. It was found that cellulose was readily degraded to humins and unknown by-products in water medium at 523 K without any catalyst. The hot water might contribute to the reactions, because a great number of protons will be formed in situ at elevated temperatures26, and function as Brønsted acids to catalyze the hydrolysis of cellulose to glucose and oligomers,as well as their further dehydration and/or polymerization. The presence of H2WO4enhanced the transformation of cellulose,but very trace amount of C2compounds such as ethylene glycol was obtained. Pt/ZrO2alone primarily facilitated the hydrogenation of cellulose to sorbitol. Once H2WO4was combined with Pt/ZrO2, ethanol could be immediately generated.Moreover, the yield of ethanol increased and that of sorbitol decreased with increasing the amount of H2WO4, which indicated that H2WO4worked for the C-C bond cleavage. The investigation on Pt loading amounts demonstrated that ethylene glycol was largely formed when Pt loading was less than 2% in the presence of H2WO4. An increase in Pt loading amount led to an increase in ethanol yield and a decrease in the ethylene glycol yield, indicating Pt nanoparticles as active sites for C-O bond scission.
Further studies pointed out that the valence of Pt nanoparticles had a significant effect on the hydrogenolysis of C-O bonds in cellulose16. A Pt/ZrO2catalyst, composed of 30% Pt0and 70% Pt2+, would provide ethanol with a yield of 32% at 523 K in the presence of H2WO4. Compared to this Pt/ZrO2(30% Pt0and 70% Pt2+) catalyst, both Pt/ZrO2-c (30% Pt4+and 70% Pt2+) and Pt/ZrO2-r (27% Pt2+and 73% Pt0) were less selective in the production of ethanol. It was notable that over-hydrogenolysis occurred over these catalysts, producing light alkanes such as CH4, C2H6, C3H8and C4H10. Pt/ZrO2-r catalyst exhibited the highest yields towards these gas products, followed by Pt/ZrO2and Pt/ZrO2-c, which clearly suggested that the metallic Pt0species was more active in the C-O bond cleavage than Pt4+and Pt2+species. Thus, high fraction of Pt0species enhanced the hydrogenolysis, but at the same time they also resulted in undesirable over-hydrogenolysis. Introduction of Pt2+into Pt0species could help to dilute Pt0dispersion and suppress overhydrogenolysis. As a consequence, a high selectivity of ethanol was obtained on Pt/ZrO2that had an appropriate Pt2+to Pt0ratio.
By loading Pt nanoparticles onto mesoporous WOx(Pt/WOx)and modifying with MoOxspecies, Zhang and co-workers have also obtained ethanol from one-pot transformation of cellulose in water medium17. Over this Mo/Pt/WOxcatalyst, an ethanol yield of 43.2% had been attained after conversion of cellulose at 518 K. Notably, deposition sequences of Pt and Mo had an influence on the formation of ethanol. When the Mo/Pt/WOxcatalyst was prepared by the Pt-Mo loading sequence, much higher yield of ethanol was attained than that on the catalyst prepared by a Mo-Pt sequence. Moreover, compared to physical mixture catalysts, WOx+ Mo/Pt/SiO2and WOx+ Pt/SiO2, this Mo/Pt/WOxcatalyst still showed better efficiency in ethanol formation. It is likely that the WOx, Pt, and Mo species in the catalyst generated a unique structure, and might work in a synergistic manner for the selective conversion of cellulose.Structural characterizations for Mo/Pt/WOxrevealed that coordinately unsaturated MoOxspecies were highly isolated on WOxsupport and bonding with Pt0metal, generating MoOx-Pt-WOxspecies. Such low-coordinated MoOxspecies could not only promote the adsorption of the C2intermediate (i.e., ethylene glycol), but also be able to tune the interaction of Pt and WOxby accepting the electrons of Pt. Moreover, the MoOx-Pt-WOxwould function as frustrated Lewis pairs to dissociate H2into H+and H-species, which are highly active and capable of selectively cleaving C-OH bond in ethylene glycol, a ratedetermining step for ethanol formation. The catalyst prepared by an inverse loading sequence of Pt and Mo did not form such unique MoOx-Pt-WOxstructure, therefore exhibiting low efficiency in ethanol production.
Another efficient catalyst consisting of tungsten-based compounds and hydrogenolysis metals is Ru-WOxsupported on zeolite HZSM-518. Over the Ru-WOx/HZSM-5(50) catalyst, the yield of ethanol could approach 77% from the transformation of cellulose in water at 508 K. X-ray diffraction analysis of Ru-WOx/HZSM-5 indicated that besides the highly dispersed Ru and WOxnanoparticles, Ru3W17alloy was also formed on HZSM-5 surfaces. These components in Ru-WOx/HZSM-5(50)catalyzed the cellulose transformation in a synergistic manner.WOxwas identified as catalytic centers to promote the retroaldol fragmentation of glucose, a hydrolysis product from cellulose, to glycolaldehyde intermediate through C-C bond cleavage, while Ru3W17alloy functioned as active sites to facilitate the hydrogenation of glycolaldehyde to ethylene glycol, and further transformation of ethylene glycol to ethanol.HZSM-5 played essential roles in ethanol production by supplying acidic sites in the hydrolysis of cellulose and dehydration of ethylene glycol. However, the acid sites might also catalyze undesirable side reactions such as dehydration of glucose and oligomerization of glucose. The H-ZSM-5(50) with moderate acidity (Si/Al molar ratio = 50) might minimize the side reactions while maintaining catalytic activity for desirable hydrolysis or dehydration, therefore exhibiting high ethanol selectivity. To improve the retro-aldol fragmentation and hydrogenation without increasing acidity, the introduction of additional Ru/WOxinto such Ru-WOx/HZSM-5(50) catalyst would significantly promote the ethanol yield to as high as 87.5%. Note that the catalyst deactivated due to the WOxleaching and poor hydrothermal stability of HZSM-5.
The combination of hollow HZSM-5 supported Pt(Pt@HZSM-5) and Pt/WOxhad also realized the conversion of cellulose to ethanol in water at 518 K27. Over an optimized catalyst, the ethanol yield could reach 54%. The hollow structure of HZSM-5 had more external acid sites and was more favorable for the mass diffusion than bulky ones, therefore exhibiting better activity in the dehydration of the C2intermediate ethylene glycol to ethanol. Recently, a neutral SO2supported Pd-Cu-WOxcatalyst had been reported to catalyze the transformation of cellulose into ethanol at 573 K28. Although solid acid is absent in this catalytic system, the in situ formed H+in hot water could accelerate the cellulose hydrolysis to glucose. After C-C bond cleavage of glucose over WOxspecies, the formed C2intermediate was transformed to ethylene glycerol over Pd.Eventually, the active Cu+/Cu0species catalyzed the selective C-O bond cleavage of ethylene glycol to yield ethanol.
Without the aid of tungsten compounds, a grapheneencapsulated nickel nanoparticles (Ni@C) catalyst in combination with H3PO4has been demonstrated to be active in the one-pot transformation of cellulose into ethanol in aqueous medium19. The graphene layers on Ni@C catalyst created an electronic negative surface through the electron transfer from Ni to grephene shells, and catalyzed the selective cleavage of C-OH and C-C bonds. H3PO4as a Brønsted acid was responsible for the hydrolysis of cellulose to glucose.Furthermore, H3PO4could serve as a “ligand” to coordinate with glucose through interaction between P-OH and hydroxyl groups, forming a cyclic di-ester that was a key intermediate for ethanol formation. The Ni@C-H3PO4catalytic system could afford an ethanol yield of 69.1% from the transformation of cellulose at 473 K and 6 MPa H2. In contrast, other mineral acids such as H2SO4, HCl and heteropoly acids were unable to form cyclic di-ester with glucose, and consequently, their combination with Ni@C catalyst was less effective in the ethanol synthesis.
Although several heterogeneous catalysts and catalytic systems have demonstrated high selectivity towards ethanol in one-pot conversion of cellulose, the leaching of active species(e.g., metal oxides) under hydrothermal conditions and the use of low cellulose concentration might considerably restrict the application of this one-step process. Therefore, more efficient and stable catalysts are still needed to make it feasible in economy.
3 The cleavage of two and more C-OH bonds in sugars and sugar acids
Sugars and sugar acids including glucose, erythritol, and glucaric acids have multiple hydroxyl groups. The selective removing two and even more hydroxyl groups are expected to form corresponding olefins, oxygen-reduced sugars, or dicarboxylic acid, which can serve as key monomers for the production of synthetic rubbers and polymers. For example, the cleavage of two vicinal C-OH bonds inside erythritol may lead to the formation of butanediols that are used in the polybutylene terephthalate and polyurethanethe industries. When the four C-OH bonds of glucaric acid were cleaved, adipic acid, an important monomer for nylon production, would be attained.Hydrogenolysis is a powerful strategy to cleave C-OH bonds,but it is generally applied in the cleavage of one C-O bond, or used to cut all the C-O bonds that possess similar dissociation energies in sugars, providing alkanes as the main products20. In contrast, deoxydehydration (DODH) enables the selective removal of two neighboring hydroxyls from polyols and emerges as an attractive reaction for sugars deoxygenation.Typically, the DODH reaction is applied to the selective deoxygenation of diols in a cis-form that can facilely coordinate with the catalyst active center, forming a metal-diolate intermediate (Fig. 3). After abstraction of oxygen atoms by the catalyst, the metal-diolate undergoes C-O bond cleavage,affording an olefin product and oxidized catalyst. Subsequent reduction of the oxidized catalyst with reducing agents recovers the catalyst, while hydrogenation of the olefin may produce corresponding saturated compound. Rhenium, vanadium, and molybdenum-based catalysts are able to form medium strength of metal-oxygen bonds and have enough oxophilicity to abstract oxygen from oxygenates. Hence, these transition metals have been widely used as active sites for DODH reaction29-32.

Fig. 3 DODH reaction of diols to olefins 30.
Homogeneous Re catalysts such as CH3ReO3, NH4ReO3, and HReO4have demonstrated excellent catalytic performances in the production of oxygen-reduced sugars, olefins, and dicarboxylic acids from the deoxygenation of sugars and sugar acids, but their separation and recovery remains a difficult33,34.The design of active Re-based heterogeneous catalysts for DODH reactions is a promising approach to overcome the problems in homogeneous systems, and thus is receiving ever increasing attention. ReOxnanoparticles alone or loaded on various supports such as activated carbon, TiO2, ZrO2, and zeolites are one type of DODH catalysts in the deoxygenation of vicinal cis-diols of sugars, primarily producing olefin products35-38. Because the ReOx-based catalysts follow a very similar reaction mechanism to the homogeneous Re catalyst, the redox of ReOxspecies such as Re7+/Re5+and Re6+/Re4+, play crucial roles in the DODH. However, the dispersion of ReOxspecies on catalyst surfaces is generally lower than that of the atomic-isolated homogeneous Re catalyst, making them less active as compared to CH3ReO3, NH4ReO3, and HReO4.
The modification of ReOx-based heterogeneous catalysts by a noble metal such as Au and Ag presents an effective strategy to enhance the DODH efficiency39-42. For instance, Tazawa et al.reported various ceria-supported ReOxand Au catalysts for the catalytic deoxygenation of vicinal diols using H2as the reducing agent39. When ReOx/CeO2alone was utilized for the deoxygenation of glycerol at 413 K for 2 h, an extremely low conversion (2%) was obtained. The addition of a small fraction of Au into ReOx/CeO2led to a glycerol conversion of 25%,which was ten times higher than that over ReOx/CeO2.Moreover, the selectivity to olefin products was also increased from 87% on ReOx/CeO2to 97% on ReOx-Au/CeO2. It is likely that Au nanoparticles enhanced the redox of Re species via H2activation, therefore improving the DODH activity. A decrease in the size of Au nanoparticles would increase the activity, but the selectivity of olefin product decreased, due to the occurrence of further hydrogenation. The Au nanoparticles with an appropriate size of approximate 10 nm was found to be active enough to activate H2while keeping the C=C bonds almost untouched during the DODH reaction. When this catalyst was applied to the conversion of erythritol, 1,3-butadiene with a yield of 81% could be obtained after reaction at 413 K for 60 h. There were two main byproducts: 2-butene, which was obtained from partial hydrogenation of 1,3-butadiene, and 2-buten-1,4-diol, a DODH product from the cleaving C2-OH and C3-OH bonds in erythritol. Because ReOx-Au/CeO2catalyst is highly selective in cleaving the C-OH bonds of cis-diols, it also displayed outstanding performance in the catalytic transformation of various methyl glucosides, which are facilely produced from esterification of sugars, into their corresponding oxygen-reduced sugars with retaining the original stereo-structures41. For example, after the deoxygenation of methyl α-D-mannopyranoside at 413 K in 1,4-dioxane, the ReOx-Au/CeO2catalyst could precisely catalyze the cleavage of cis-vicinal hydroxyls at C2and C3positions, providing methyl 2,3-dideoxyα-D-erythro-hex-2-eno-pyranoside with a yield of ~92% (Fig.4). Moreover, the catalytic conversion of sugars with cis-vicinal OH groups such as methyl α-L-rhamnopyranoside, methyl β-L-arabinopyranoside, and methyl β-D-galactopyranoside could lead to the formation of corresponding unsaturated sugars with high selectivities (84%-93%). Clearly, the ReOx-Au/CeO2-catalyzed DODH reaction has offered a simple and effective way to create oxygen-reduced unsaturated sugars that generally produced from conventional synthetic methods requiring multiple steps (5-7 steps)41.
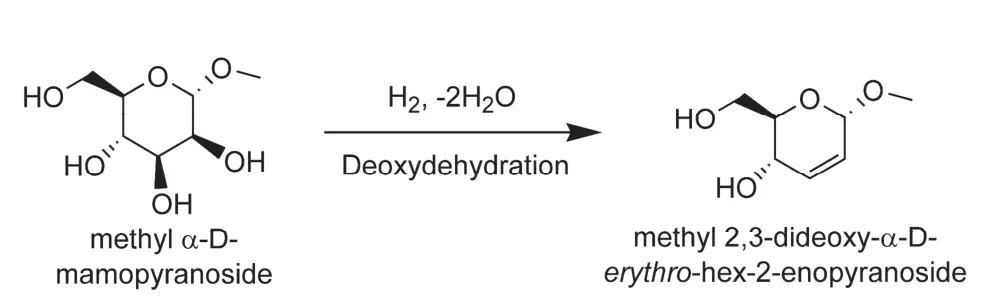
Fig. 4 Deoxygenation of methyl glucosides to oxygen-reduced sugars 41.
When ReOx/CeO2was modified by Pd instead of Au or Ag,the obtained ReOx-Pd/CeO2also demonstrated superior catalytic performance in the deoxygenation of cis-diols, but the major products shifted from the olefins to corresponding saturated alcohols43-45. For example, the deoxygenation of erytritol would generate 1,2-butanediol rather than 2-buten-1,4-diol, while the conversion of methyl α-D-mannopyranoside mainly afforded the saturated dideoxy sugar, methyl α-D-2,3-dideoxymannopyranoside. These observations indicated that the ReOx-Pd/CeO2had enhanced hydrogenation and deoxygenation at the same time. Detailed reaction mechanism studies on noble metal modified ReOxcatalysts disclosed that the DODH reaction proceeded via three key steps: (i) the H2-accelerated reduction of high oxidation state of Re such as Re6+to a low-coordinated and highly active Re such as Re4+, (ii) the coordination of cis-diols with the active Re to yield Re-diolate intermediate, and (iii)subsequent fracture of the C-O bonds inside the Re-diolate to give one olefin product and oxidized Re species. After reduction by H2, the oxidized Re species could return to the active lowoxidation state, participating in the next catalytic cycle. Because noble metals including Au, Ag, and Pd were able to activate H2,they might promote the DODH reaction by accelerating the reduction of high-valent Re species. Compared to Au and Ag, Pd exhibits higher activity in the hydrogen activation, and thus it is reasonable that ReOx-Pd/CeO2tends to afford saturated products via an easy hydrogenation of unsaturated DODH products (Fig.5).
Most Au and Pd modified ReOx/CeO2catalysts have succeeded in the deoxygenation of various sugars to olefins or oxygen-reduced sugars, but they are only selective in the cleavage of cis-diols. The catalytic conversion of sugars or sugar acids with trans-diols remains a difficult. A typical example is the synthesis of adipic acid from deoxygenation of glucaric acid,a dicarboxylic acid containing both trans-diols and cis-diols. In 2017, Toste and co-workers reported a catalytic system consisting of potassium perrhenate (KReO4), Pd/C and H3PO4in the deoxygenation of glucaric acid-3,6-lactone to methyl adipate in methanol medium46. Because the two pairs of diols in glucaric acid-3,6-lactone are favorable for the formation cisconfiguration, they can be removed via tandem reactions including the first DODH of diols in the lactone ring,transesterification to open the ring, and subsequent DODH reaction (Fig. 6). In this catalytic system, KReO4was primarily responsible for DODH, while Pd/C acted as a co-catalyst to facilitate DODH and hydrogenation. Furthermore, the additives including H3PO4and activated carbon also played roles in the deoxygenation by promoting the ring-opening of intermediates and adsorbing undesirable products that might poison the active sites. Under the reaction conditions of 423 K and 18 h, this combined catalytic system offered dimethyl adipate with a yield of 88%.

Fig. 5 A proposed mechanism for the ReOx-Au and ReOx-Pd catalyzed DODH reactions 43.
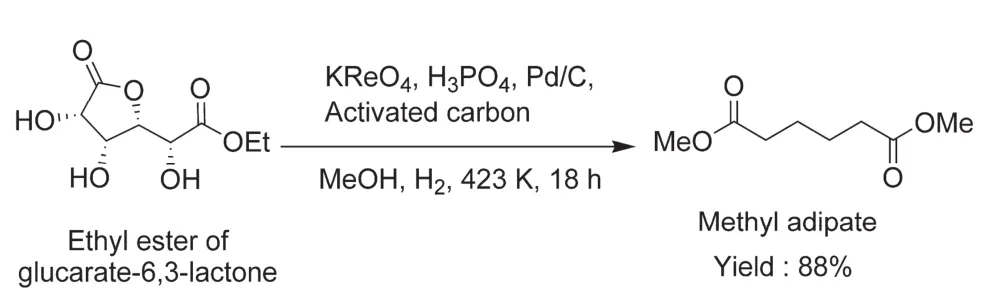
Fig. 6 Conversion of glucaric acid-3,6-lactone to methyl adipate 46.
By using zirconia supported ReOxas a heterogeneous catalyst,Lin et al. have obtained adipic acid from the catalytic deoxygenation of glucaric acid-1,4-lactone in n-butanol under flow N247. At a reaction temperature of 393 K, ReOx/ZrO2could offer two DODH products with an overall yield of 93%. One is a linear ester bearing conjugated alkenes (dibutyl hexa-2,4-dienedioate) and the other is a lactone ester containing one hydroxyl group and one alkene (Fig. 7). The presence of lactone product suggested the occurrence of incomplete deoxygenation.Notably, this lactone product could not undergo further ringopening and deoxygenation by increasing the reaction time. A possible reason is that the rigid conjugated structure formed between alkene and carbonyl groups inhibited the ring-opening reaction. When the unsaturated lactone product was reduced to a saturated intermediate by Pd/C catalyzed-hydrogenation, the further ring-opening and deoxygenation became easy over the ReOx/ZrO2catalyst (Fig. 7). Because Pd/C negatively influenced the catalytic behavior of ReOx/ZrO2, it is necessary to conduct the ReOx/ZrO2-catalyzed DODH and Pd/C-promoted hydrogenation reactions separately. Four consecutive DODH and hydrogenation reactions finally resulted in dibutyl adipate with a yield of 82%. Notably, alternatively conductions of the DODH and hydrogenation under different atmosphere, and tedious separation of the ReOx/ZrO2and Pd/C from the reaction medium may markedly increase the cost of such catalytic system, thus restricting its practical application.
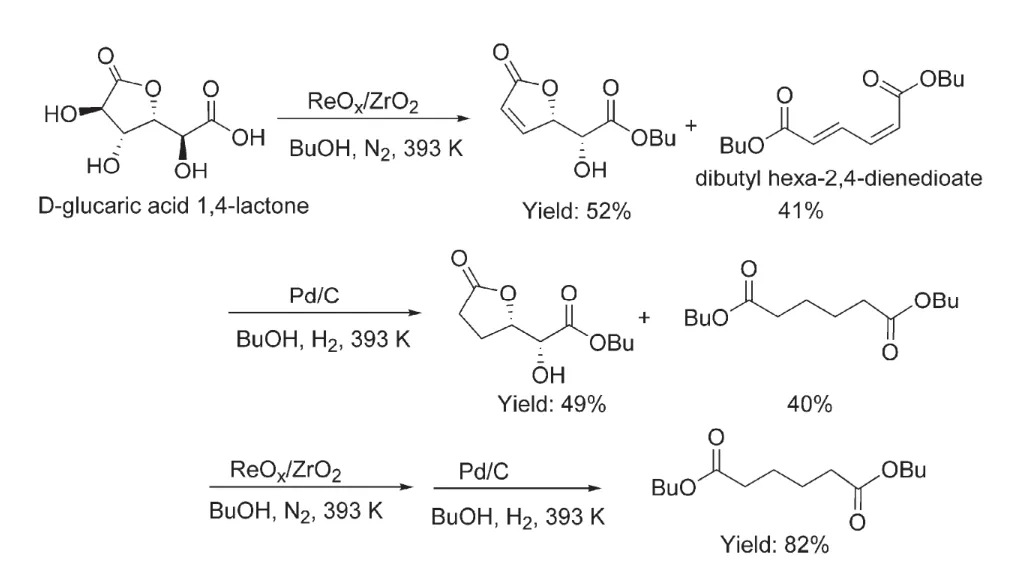
Fig. 7 Synthesis of dibutyl adipate from catalytic deoxygenation of D-glucaric acid 1,4-lactone through a combination of ReOx/ZrO2-catalyzed DODH and Pd/C-promoted hydrogenation 47.
Recently, a bifunctional catalyst constructed by loading Pd and ReOxnanoparticles on activated carbon (Pd-ReOx/AC) has been reported to achieve the adipic acid production from one-pot deoxygenation of potassium glucarate, which was a salt of glucaric acid obtained by glucose oxidation under alkaline conditions48. It was found that ReOx/AC alone could catalyze the DODH reaction of potassium glucarate at 383 K in methanol medium in H2, but mainly afforded partial DODH products, that is, one pair of cis-diols of glucarate were removed. Despite that the formation of dimethyl adipate also occurred over ReOx/AC,the yield was less than 5%. Pd/C catalyst displayed almost no activity in the potassium glucarate conversion. When Pd-ReOx/AC catalyst was used, a near-quantitative yield of dimethyl adipate (95%) could be obtained, strongly suggesting that ReOxand Pd species catalyzed the DODH reaction synergistically. Moreover, Pd-ReOx/AC was also superior to various combination catalysts that were composed of Pd/C and homogeneous rhenium catalysts (e.g., CH3ReO3, KReO4,HReO4, and NH4ReO4). Mechanism studies indicated that the Re with a high oxidation state such as Re6+was identified as active sites in deoxygenation, whereas metallic Pd catalyzed the saturation of olefin intermediates. Moreover, the Pd nanoparticles also promoted the redox of Re6+/Re4+species in DODH reaction by accelerating H2activation. In addition, the presence of Pd nanoparticles in Pd-ReOx/AC significantly had facilitated the dispersion of ReOxspecies, which was confirmed by transmission electron microscopy analysis. Theoretical computations suggested the binuclear Re-O-Re sites on catalyst surfaces as active centers for the DODH reaction. Compared to single Re site such as a homogeneous Re compound, the binuclear Re sites have more space to coordinate with two vicinal -OH groups, and thus either trans-diols or cis-diols can facilely bond and coordinate with the binuclear Re atoms,forming key Re-diolate complexes. As a consequence, the onepot deoxygenation of glucaric acid can smoothly proceed over the Pd-ReOx/AC catalyst.
4 Deoxygenation of furan compounds
5-Hydroxymethyl furfural (HMF), a typical cellulose-derived furan compound, has three different oxygen-containing groups,i.e., carbonyl (C=O), hydroxyl (C-OH), and ether(C-O-C). Because of these abundant functional groups, HMF can be transformed into various valuable chemicals through selectively cleaving C-O bonds. For example, deoxygenation of carbonyl/hydroxyl groups enables the formation of 2,5-dimethylfuran (DMF) that has much higher energy densities than ethanol (30 vs. 21 kJ·cm-3), and can be served as a potential gasoline additive49-54. The hydrogenolysis of the ether bond(i.e., C-O-C) inside HMF will open the furan ring, resulting in a linear product such as 1,6-HD. Therefore, the precise activation and cleavage of particular C-O bond is of crucial to achieve the catalytic conversion of HMF and its derivatives to valuable chemicals.
4.1 C=O/C-OH bonds cleavage
The synthesis of DMF from HMF generally includes the hydrogenation of carbonyl group into a hydroxyl group and immediate hydrogenolysis of the hydroxyl groups (Fig. 8). Since transition metals (e.g., Pd, Ru, Pt, Cu, Co, and Ni) and metal carbides (e.g., W2C, α-MoC, and Mo2C) can facilitate hydrogenation and hydrogenolysis, they are extensively used in the catalytic hydrodeoxygenation of HMF and its derivatives49-54.However, monometallic metal-based catalysts are also capable of catalyzing the hydrogenation of conjugated carbon-carbon double bonds in furan rings, and even may promote ring-opening reactions. Therefore, much effort has been devoted to enhancing the deoxygenation selectivity by tuning the reaction parameters including reducing agents and reaction medium, or modifying the properties of supports55-61.

Fig. 8 The formation of DMF from HMF conversion 49.
For example, a Pd/C catalyst in combination with formic acid and H2SO4could afford DMF with a yield of more than 95% from the conversion of HMF at 343 K55. During the reaction,formic acid played dual roles in deoxygenation: one was providing hydrogen source for hydrogenation, and the other was acting as a reagent to form ester intermediate with HMF. After the H2SO4-promoted esterification of HMF and formic acid, the C-O bonds in ester intermediate became easily cleaved by Pd/C under mild conditions, forming DMF with a high selectivity.When a special carbon support was designed to load active metals such as Ru and Co, the formation of DMF could also be enhanced59,60. The polyphenylene (PPhen)-supported Ru nanoparticles has demonstrated a DMF yield of 92% from HMF reaction at 433 K and a Ru/HMF molar ratio of 2.8%. PPhen was mainly composed of tetra-substituted benzene groups and phenylene. The abundant sp2carbons facilitated the interaction of PPhen and HMF via π-π stacking. Because of such interaction, HMF molecules were isolation, avoiding the formation of intermolecular hemiacetal, which might generate unwanted humins as side products. Moreover, PPhen also confined the Ru clusters in the framework by metal-PPhen interaction, which favored the C-OH bond hydrogenolysis without hydrogenation of the C=C or C=O double bonds.Thus, high selectivity of DMF was attained62. Additionally, the solvent could also exert effects on the selecitivity of DMF over a Ru/C catalyst61. In a miscible mixture solvent composed of an ionic liquid ([BMIm]Cl) and a dipolar organic such as CH3CN,n-butanol, and i-propanol, very low DMF yields of 11%-15% were attained after reaction at 403 K for 30 min. When tetrahydrofuran (THF) was mixed with [BMIm]Cl, the DMF yield could be enhanced to ~50%. This enhancement was probably ascribed to the formation of biphasic between ionic liquid and THF. The cage effect of ionic liquid improved the deoxygenation of HMF, whereas THF timely extracted the DMF from ionic liquid and prevented it from further reaction,therefore leading to a high selectivity of DMF.
Another strategy to improve the deoxygenation of HMF is design of bimetallic catalysts that integrate multiple functions of two different metals. One typical example is the combination of an oxophilic metal such as Fe with an active metal such as Pd,Pt, Ni, and Cu. Oxophilic metals are expected to adsorb the oxygen-containing groups such as hydroxymethyl and carbonyl,whereas active metals may activate hydrogen, and catalyze the hydrogenation and hydrogenolysis of the adsorbed C=O/C-O bonds.
For example, Yuan and co-workers constructed a Fe-Ni alloy on carbon nanaotube, and performed the hydrodeoxygenation of HMF63. The Fe-Ni/CNT catalyst with a Fe/Ni ratio of 1/2 produced DMF at a yield up to 91% under an optimized reaction condition of 3 h at 473 K and H2of 3 MPa. In the absence of Fe,Ni catalyst afforded only 46% of DMF yield at identical conditions. Similar phenomena have been observed on the Fe-Cu bimetallic catalyst64. At a Fe/Cu ratio of zero, the monometallic Cu catalyst gave HMF conversion of 80% and DMF selectivity of 59% at 443 K. At an Fe/Cu ratio of 2, the HMF conversion and MF selectivity were remarkably raised to 97%, and 93%, respectively. At the same time, furan alcohol and furan that formed on Cu via decarbonylation were considerably suppressed over the Cu-Fe catalyst. X-ray diffraction analysis indicated that spinel CuFe2O4was formed as the introduction of Fe into Cu. Moreover, Fe-Cu metallic catalyst gained strong Lewis acidity at high Fe/Cu ratios. It is likely that the enhanced Lewis acidity as well as the oxophilic nature of Fe facilitated the adsorption and activation of carbonyl group inside HMF. As a result, the reduction of C=O became much easy. Furthermore,CuO/Cu species provided Brønsted acid sites together with metallic Cu to catalyze the subsequent dehydration/hydrogenation of hydroxymethyl groups, giving deoxygenation product64.
Bimetallic catalysts combining active noble metals (e.g., Ru,Pt, and Pd) with other non-noble metals (e.g., Cu, Re, Zn, and Co) have also been reported for the catalytic deoxygenation of HMF58,65-79. In these bimetallic catalysts, one metal, commonly noble metal provides active centers to catalyze hydrogenolysis or hydrogenation, while the other metal either modifies the properties of the active centers or assists the hydrogenolysis and hydrogenation by tuning the properties of the catalysts for adsorption or activation of the oxygen-containing groups. For instance, Gorte and co-workers prepared one Pt3Co2alloy and one Pt/C catalyst, and investigated their catalytic performances for the hydrodeoxygenation of HMF at 433 K70. The Pt/C was found to demonstrate high active in both hydrogenation and hydrogenolysis, but it tended to catalyze the overhydrogenolysis or over-hydrogenation of the target product(DMF) and HMF into saturated or ring-opening by-products(e.g., hexanedione, hexanone, and hexanol). Thus, the DMF yield only researched 41% over Pt/C. Under the same reaction conditions, Pt3Co2alloy catalyst dramatically suppressed the further transformation of DMF and afforded DMF with a yield up to 98%. This Pt3Co2catalyst was composed of a core with Ptrich phase and a surface monolayer with CoOxspecies.Theoretical calculations revealed that the CoOxmonolayer on the metallic core surfaces could provide active sites to perform the hydrodeoxygenation. Importantly, it could weakly interact with the furan rings of HMF and DMF, avoiding various undesirable reactions such as ring-opening and overhydrogenation. In contrast, metallic Pt favored strong interaction with the DMF furanic ring, and facilitated the hydrogenation of conjugated carbon-carbon double bonds and ring-opening reactions. Thus, the inhibition of further transformation of DMF by the alloy catalyst ensured its excellent selectivity to DMF.
Recently, a PdCu nanoalloy has been fabricated to catalyze the transformation of HMF to DMF at ambient temperature80. It was clarified that the reaction path and product distribution were largely determined by the crystallographic phases of the PdCu catalysts. Body-centered cubic (BCC) PdCu nanoalloy loaded on activated carbon (PdCu/AC-BCC) showed exceptional performance in the formation of DMF, giving a DMF yield of 93.6% at 303 K, whereas the face-centered cubic (FCC) PdCu nanoalloy (PdCu/AC-FCC) catalyst primarily catalyzed undesirable hydrogenation of conjugated carbon-carbon double bonds in furan ring, giving DMF with a yield of only 9% under the same reaction conditions. Density functional theory (DFT)studies on two PdCu alloy models had unraveled the origin of selectivity difference over the PdCu nanoalloy with different crystalline-phases. Two types of adsorption configurations for HMF and its product were identified on the PdCu surfaces: σadsorptions and η-adsorptions. In the η-adsorption modes, HMF or its derivatives interacted with PdCu surfaces through the oxygenated pendant functional (e.g., hydroxyl and aldehyde),whereas the furan ring preferentially lied on PdCu surfaces in σadsorption mode (Fig. 9). The adsorption energies of HMF on PdCu-FCC (100) and PdCu-BCC (111) surfaces through σadsorption configuration were found to be stronger than the ηadsorption mode, and they were the estimated to be -0.71 and-1.00 eV, respectively. In the σ-adsorption mode, both aldehyde group and furan ring in HMF could be activated, but the hydrogenation of aldehyde group preferentially occurred and produced 2,5-bis(hydroxymethyl)furan (DHMF). Once the DHMF was formed, the η adsorption mode became favored on the surfaces of PdCu-BCC, keeping the furan ring far from the surface. In contrast, DHMF was prone to adsorb in σ-adsorption mode on PdCu-FCC surfaces, which was 0.43 eV lower than in the η adsorption configuration. Consequently, the FCC crystalline phase facilitated the activation of furan ring, and favored the hydridization of furan Csp2to Csp3by generating carbon-metal bonds. Such interaction between PdCu-FCC (100)surfaces and furan rings was beneficial to the hydrogenation of furan rings, interpreting the high selectivity towards ringsaturated products on PdCu-FCC. In addition, the adsorption energy of DMF on PdCu-BCC (100) surface was weaker compared to that on PdCu-FCC (100) (-0.33 eV vs. -0.45 eV).This suggested much easier desorption of DMF from PdCu-BCC(100), avoiding deep reactions. Thus, the difference in adsorption behaviors of HMF, intermediates and DMF on PdCu-FCC and PdCu-BCC surfaces determined the ring hydrogenation reactivity, and finally controlled over the product selectivity.
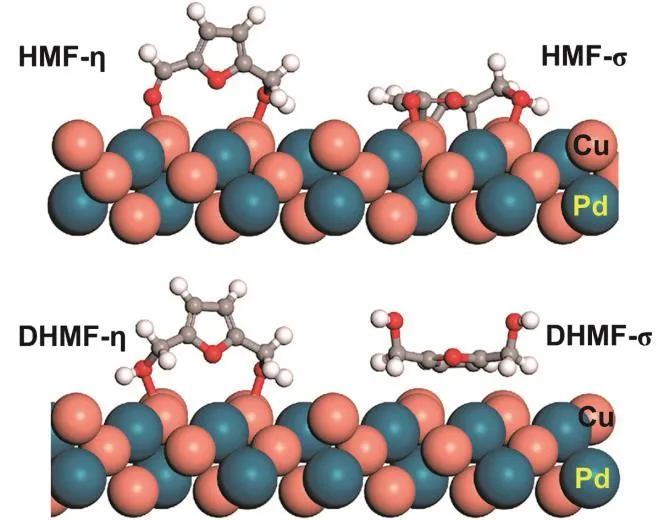
Fig. 9 Adsorption of DHMF and HMF on PdCu surfaces with η and σ configurations 80.
4.2 C-O-C bonds cleavage
When hydrogenolysis is applied to cleaving the C-O-C ether bonds, HMF can be transformed to ring-opening product such as 1,6-HD. Generally, the formation of 1,6-HD include three tandem reactions: (1) the initial hydrogenation of furan ring inside HMF and carbonyl groups generates 2,5-furandimethanol or 2,5-tetrahydrofuran-dimethanol (THFDM) intermediates; (2)selectively opening the five-membered ring of THFDM and 2,5-furandimethanol via hydrogenolysis to form 1,2,6-hexanetriol(1,2,6-HT); (3) subsequent deoxygenation of 1,2,6-HT affords 1,6-HD (Fig. 10). Each step involves the activation of different chemical bonds such as the carbon-carbon double bonds in the first step, ether bonds in the second, and the C-OH bond in the third. Thus, one strategy to achieve high efficiency in production of 1,6-HD is to develop an appropriate catalyst for each reaction and perform the reaction separately. For example, Vries, Heeres and co-workers reported that Raney-Ni afforded THFDM with a quantitative yield from the hydrogenation of HMF at 373 K,while a Rh-ReOx/SiO2catalyst combined with a Brønsted acid enabled the selective production of 1,6-HD from the conversion of THFDM at 393 K in aqueous solution81. The overall yield of 1,6-HD reached 86%. However, Rh-ReOx/SiO2catalyst may undergo active site-leaching, because the SiO2is highly soluble under hydrothermal conditions. Thus, more stable and efficient catalysts or catalytic systems are highly desirable in the selective conversion of THFDM82-85or directly transformation of HMF into 1,6-HD.

Fig. 10 Key steps in the transformation of HMF into 1,6-HD 81.
Through loading Pt nanoparticles and tungsten oxides onto titanium oxide, Dumesic, Huber and co-workers obtained a bifunctional catalyst and investigated its catalytic performance for the hydrogenolysis of THFDM84,85. They obtained a 1,6-HD yield of 70% on the Pt-WOx/TiO2catalyst after reaction of THFDM at 433 K in H2. Moreover, a combination catalyst by physically mixing WOx/TiO2and Pt/TiO2showed similar activity as Pt-WOx/TiO2, but the mixture of WOx/TiO2and Pt/C or Pt/Al2O3had almost no activity under the same reaction conditions. This suggested that metallic Pt nanoparticles should be deposited on metal oxides (e.g., TiO2) that were reducible,when Pt and WOxspecies had no contact. Further mechanism studies unraveled that the synergistic catalysis between Pt metal,W5+/W6+, and TiO2contributed to the outstanding performance of Pt-WOx/TiO2. Initially, H2adsorbed and dissociated into hydrogen atoms over Pt nanoparticles. Then, these hydrogen atoms travelled to TiO2sites via H+and e-pair. Because Ti4+could be reduced to Ti3+by electrons, the protons (H+) diffused with electrons on TiO2surfaces assisted by Ti4+/Ti3+pairs. After spillover to WOxsites, the hydrogen atoms facilely reduced the W6+to W5+, and created plenty of W-OH sites. Such W-OH sites functioned as Brønsted acids, catalyzing the dehydration of THFDM into carbenium ions. Upon hydrogenation over Pt metal, these carbenium ions were facilely transformed into 1,6-HD.
Another strategy for 1,6-HD production is the direct one-pot conversion of HMF over multifunctional catalysts or combined catalysts86,87. In 2016, Zhang, Zheng and co-workers combined Ir-ReOx/SiO2and Pd/SiO2catalysts and investigated their catalytic performance in HMF conversion in a fix-bed reactor86.The Pd/SiO2was loaded in the upper layer to promote initial HMF to THFDM transformation, and the formed THFDM underwent further hydrogenolysis to offer 1,6-HD over Ir-ReOx/SiO2which was placed at the bottom layer. After reaction in 7 MPa H2at 373 K, the combined catalysts could provide 1,6-HD with a yield of 57.8 %. However, the 1,6-HD yield was only 14.6% on the Pd-Ir-ReOx/SiO2catalyst that consisted of the same amounts of Pd, Ir and ReOxspecies as the combination catalysts.In addition, high pressure of H2was favorable for the 1,6-HD formation. It was probably that high pressures facilitated the H2adsorption on metallic active sites, which could reduce the coverage of 1,6-HD on the catalyst surfaces. Thus, the overhydrogenolysis of 1,6-HD to by-products such as hexane and hexanol was substantially inhibited, leading to a high selectivity of 1,6-HD.
Similar to the synthesis of 1,6-HD, the cleavage of ether bond inside 2,5-furfurandicarboxylic acid (FDCA), a compound from HMF oxidation, can yield adipic acid. Hydrogenolysis is powerful in the furan ring-opening, but it can result in overhydrogenation or hydrogenolysis of the COOH groups as well,producing undesirable by-products. Thus, efficient catalysts need to selectively cleave the ether bonds while keeping carboxylic groups intact. In 2010, Boussie et al. reported a halogen-assisted-Pt/SiO2catalytic system to transform FDCA into adipic acid88. The reaction was performed via two-steps: (1)hydrogenation of FDCA over Pd/SiO2catalyst to obtain a key intermediate tetrahydrofuran-2,5-dicarboxylic acid (THFDCA)in acid medium (e.g., CH3COOH), and (2) hydrogenolysis of THFDCA to adipic acid on Pd/SiO2in the presence of HI. Pd was probably responsible for the hydrogenation of conjugated carbon-carbon double bonds in the furan ring to form THFDCA.The halogen and acid medium might play dual roles in the subsequent conversion of THFDCA to adipic acid. They not only promoted the furan ring-opening via protonation of the ring oxygen in THFDCA, but also facilitated the subsequent dehydration of hydroxyl groups in the ring-opening intermediate. After consecutive reaction of FDCA over Pd/SiO2at 413 K, and then reaction at 433 K with addition of 0.2 mol·L-1HI, the overall yield of adipic acid could reach 88%88.Moreover, the synthesis of adipic acid from THFDCA transformation could also be achieved in a metal-free catalytic system consisting of propionic acid, H2, and HI89,90. Notably,the co-presence of corrosive organic acids and halogen is a great challenge for the stability of metal catalyst and reactors. To avoid the use of organic acids, an iodide-containing ionic liquid,[MIM(CH2)4SO3H]I (MIM = methylimidazolium) has recently been designed to convert THFDCA91. The [MIM(CH2)4SO3H]I delivered an almost 99% yield of adipic acid at 453 K, and also enabled simple product isolation with high purity. However, the recycled ionic liquid needed to be treated with HI solution to recover activity, making the catalytic system less environmental benign.
Catalytic transformation of FDCA in aqueous solution is an appealing approach to synthesize adipic acid, and the reaction requires noble metal-based bifunctional catalysts that can catalyze the hydrogenation of conjugated carbon-carbon double bonds and C-O-C bond cleavage92,93. Asano et al. prepared a Pt-MoOx/TiO2catalyst by sequential impregnation method,and investigated the hydrogenolysis of FDCA in water92. It was clarified that the Pt and MoOxspecies worked synergistically to cleave the ether bonds at the Pt-Mo interface. Pt first activated and dissociated H2to cut a C-O bond inside the FDCA furan ring, whereas the neighboring MoOxspecies rapidly abstracted the ether oxygen atom by donating two electrons, resulting in another C-O bond cleavage. Then, the oxidized Mo species recovered activity upon a simple reduction by H2and participated in the next catalytic cycle. The Pt-MoOx/TiO2catalyst afforded adipic acid with a yield of 21% at 473 K. Due to the leaching of MoOxspecies in water medium, Pt-MoOx/TiO2deactivated.
By using water-tolerant niobic acid (Nb2O5·xH2O) as a Brønsted solid acid, Wang and co-workers have constructed a bifunctional catalyst Pt/Nb2O5·xH2O to investigate the hydrogenolysis of FDCA in H2O93. Compared to the Pt nanoparticles loaded on H-ZSM-5, SiO2, TiO2, and Al2O3, the Pt/Nb2O5·xH2O catalyst showed 2-6 times higher yield of adipic acid at a temperature of 473 K and H2pressure of 3 MPa.Moreover, Pt/Nb2O5·xH2O was much more stable and efficient than Pt-MoOx/TiO2that had been reported in the FDCA transformation92. After two recycling uses, the adipic yield over Pt-MoOx/TiO2was decreased from 22% to 18%. In stark contrast, Pt/Nb2O5·xH2O catalyst offered adipic acid with a yield of 38%. Furthermore, the adipic acid yield could be maintained in the five repeated runs. Kinetic studies on Pt/Nb2O5·xH2O indicated that FDCA was first converted to THFDCA through the hydrogenation of unsaturated furan ring. Then, the ring was opened by cleaving the ether bond, and formed 2-hydroxyadipic acid. Upon dehydration/hydrogenation or hydrogenolysis, the hydroxyl group of 2-hydroxyadipic acid was removed, providing adipic acid (Fig. 11). Metallic Pt species functioned as active sites for H2activation, and catalyzed the C-O-C bonds cleavage and hydrogenation of carbon-carbon double bonds.Nb2O5·xH2O participated in the C-OH bond cleavage via the acid-catalyzed dehydration. Thus, the combination of Pt and Nb2O5·xH2O facilitated the formation of adipic acid. It should be mentioned that over-hydrogenation and/or decarboxylation of carboxyl groups occurred over the catalyst, producing byproducts such as CO2, hydroxycaproic acid and 5,6-dihydroxyhexanoic acid. As a consequence, rational design of efficient and stable catalysts for the selectively cleaving ethers bond inside FDCA remains an urgent direction in the future.

Fig. 11 A proposed reaction pathway for Pt/Nb2O5·xH2O-catalyzed conversion of FDCA to adipic acid.
5 Conclusions
Efficient production of chemicals from renewable biomass, in particular cellulose, can help to build a green and sustainable chemical society and reduce our heavy dependence on fossil resources. Because of the over-functionalized oxygencontaining groups inside cellulose, it is essential to selectively remove particular oxygen atoms for the production of chemicals that can meet the requirements of current chemical industries. In the past decades, an array of valuable chemicals such as ethanol,DMF, 1,6-HD, and adipic acid have been synthesized from the catalytic deoxygenation of cellulose and its derived platforms.The deoxygenation strategies and catalyst types are found to play determining roles in the product distribution and deoxygenation efficiency.
Bifunctional catalysts that combine a noble metal (e.g., Ru and Pt) and a tungsten compound (e.g., H2WO4and WOx) have demonstrated excellent performance in one-pot transformation of cellulose into ethanol. Tungsten compounds catalyzed retroaldol reaction to break particular C-C bonds, while noble metals facilitated the hydrogenolysis of C-OH bond. The cooperation of them in one catalytic system enables the selective formation of ethanol. Despite efficient, the tungsten-containing catalysts may undergo severe W-leaching under hydrothermal conditions, significantly limiting its practical application. Thus,design of tungsten-free and more stable bifunctional catalysts remains a necessary direction for ethanol synthesis.
DODH reaction as an effective strategy to remove neighboring cis-diols has been explored to cleave two or more C-OH bonds inside cellulose-derived sugars and sugar acids.Re-based compounds are widely used DODH catalysts and show superior performance in the transformation of glucose, methyl glucosides and erythritol into corresponding olefins or oxygenreduced sugars. In typical DODH reactions, a key step is the interaction of mononuclear Re with two hydroxyl groups,forming a Re-diolate intermediate which might yield olefins via C-O bond cleavage. Because cis-diols have a more favorable configuration to interact with mononuclear Re than trans-diols,the conventional Re-catalyzed DODH reaction is applicable to the deoxygenation of cis-diols in sugars, but not feasible for the glucaric acid that has trans-diols. A bifunctional catalyst consisting of binuclear Re-O-Re species and Pd nanoparticles could achieve outstanding performance in the deoxygenation of glucaric acid to adipic acid. The easy coordination of binuclear Re-O-Re with either trans- or cis-diols and Pd-promoted redox of ReOxspecies are probably the main reasons for the high efficiency in deoxygenation. Notably, the Re-based bifunctional catalysts generally remove pairs of hydroxyl groups. When more odd numbers of hydroxyl groups inside biomass are expected to be removed, it will be a challenge for the Re-based bifunctional catalysts. Thus, development of novel multifunctional catalysts that integrate various functions such as DODH, hydrogenolysis and dehydration is desirable.
The deoxygenation of HMF can lead to the formation of either furanic compounds (e.g., DMF) or linear products (e.g., adipic acid and 1,6-HD), which depend on the bond cleavage occurring at either C-O-C or C-OH/C=O bond. Monometallic catalysts such as Pt, Pd, and Ni generally exhibit high activity in hydrogenolysis of C-OH/C=O bonds, but they may accelerate undesirable hydrogenation of furan ring to saturated compounds as well. To enhance the selectivity in C-OH/C=O bond cleavage, an effective method is the introduction of a second metal into the active metal. With appropriate electronic properties and preferential adsorption ability for C-OH/C=O bonds, bimetalltic catalysts enable selective deoxygenation of HMF to DMF. For the transformation of HMF into 1,6-HD, Pt catalysts modified by tungsten or rhenium oxides are highly effective. Brønsted acid sites generated from W-OH or Re-OH species are identified as the active centers for the C-O bond cleavage. When water-tolerant Brønsted acid Nb2O5·xH2O was combined with Pt nanoparticles, the C-O-C bonds inside FDCA could be cleaved, and produced adipic acid. However, at the same time, over-hydrogenation and decarboxylation of carboxyl groups also occurred, significantly restricting the selectivity of adipic acid. As a consequence, efficient strategies and catalysts are still in urgent needs to achieve the selective deoxygenation of the oxygen inside furan ring while keeping the carboxylic groups untouched.
Moreover, the stability of the deoxygenation catalysts is also of importance under hydrothermal conditions, since the water is not only a green solvent, but also can act as a Brønsted acid to participate in the hydrodeoxygenation. The use of water-tolerant materials to support or confine active sites may be a promising strategy to enhance the stability. Additionally, the fabrication of efficient catalysts that are workable under mild conditions (e.g.,room temperature or < 373 K) is also beneficial to the catalyst stability.
猜你喜欢
无机化学学报(2024年1期)2024-01-20 03:55:50
化工职业技术教育(2021年5期)2021-11-09 03:10:40
化工职业技术教育(2021年1期)2021-03-15 06:57:44
中国洗涤用品工业(2020年2期)2020-05-16 11:28:40
Communications in Theoretical Physics(2017年6期)2017-05-12 08:52:41
中国洗涤用品工业(2017年2期)2017-04-16 05:07:39
爱你(2017年10期)2017-04-14 11:21:51
文学教育(2016年33期)2016-08-22 12:58:26
World Journal of Integrated Traditional and Western Medicine(2016年4期)2016-03-28 02:08:04
化工学报(2016年3期)2016-03-14 08:37:00
