
糖皮质激素诱导细胞凋亡的研究进展
2022-10-14 01:36项晓洪胡景伟
国际医药卫生导报 2022年20期
项晓洪 胡景伟
内蒙古医科大学赤峰临床医学院儿内科,赤峰 024000
糖皮质激素(glucocorticoids,GC)是肾上腺皮质分泌的一类甾体激素,也可人工合成,是临床治疗中主要用于抑制免疫应答、抗炎、抗休克的药物[1]。但GC 使用也受到不良反应的限制,例如骨质疏松、高血糖、心血管疾病、加重神经损伤等。其机制可能是由GC诱导细胞凋亡引起。在GC敏感细胞中,细胞凋亡通过糖皮质激素受体(glucocorticoid receptors,GR)来诱导的。然而,在不同组织细胞中,GC 诱导细胞凋亡信号通路机制不尽相同。本文将从各个器官系统层面讲述其诱导凋亡信号传导通路的研究成果,为临床使用GC提供一定的理论基础。
GC诱导细胞凋亡的一般机制
在GC敏感细胞中,细胞凋亡和其他细胞效应是通过激活GR 来诱导的GR 是一种类固醇激素受体,与配体结合后移位到细胞核,发挥多种基因组和非基因组的效应。GR 两种主要的亚型:GRα 和GRβ。GRα 具有诱导细胞凋亡。虽然GRβ无法结合GC,但是其可通过竞争共调节因子或形成无活性的GRα/GRβ 异源二聚体抑制GRα[2]。GR 介导的基因激活对于GC诱导的细胞凋亡的启动是必不可少的,因为后者在放线菌素D 和放线菌亚胺的存在下被阻断,从而表明需要从头转录和翻译[3]。GC诱导细胞凋亡途径主要包括外在途径、内在途径及内质网应激(endoplasmic reticulum stress,ERS),但是GC 诱导的细胞凋亡的经典途径涉及到外在和内在的凋亡途径的激活。具体见图1。
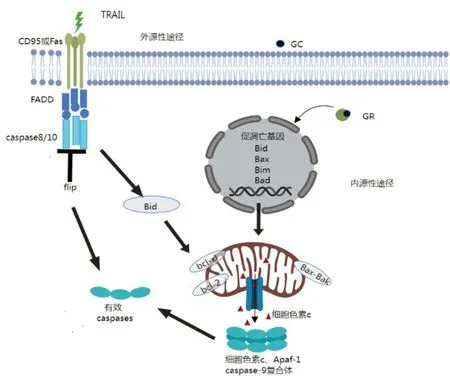
图1 GC诱导细胞凋亡的内源性及外源性凋亡途径
1、内源性途径
内源性途径是GR 信号诱导的和线粒体依赖地对各种内源性刺激(如紫外线照射和饥饿)做出的反应。Bcl2家族成员中抗凋亡成员:Bcl-2、Bcl-XL、Bcl-w,促凋亡成员:Bax、Bcl-Xs、Bad、Bak、Bik、Bid、Bim 等[4]。促凋亡和抗凋亡的Bcl2家族成员之间的相互作用密切调控着细胞的凋亡反应。半胱天冬酶的家族成员(caspases)与凋亡相关的凋亡启动因子和效应因子。细胞凋亡启动子:caspase-8、caspase-10、C-FLIPL、caspase-9、caspase-2;细胞凋亡效应因子:caspase-3、caspase-7、caspase-6[5]。GC 信号通过上调促凋亡蛋白Bim 的表达,激活促凋亡蛋白Bak 与Bax,从而促进Bak、Bax形成Bax/Bak寡聚体。Bax/Bak破坏线粒体膜电位,导致细胞色素c和其他凋亡蛋白释放至胞浆中。然而,这一过程遭到了抗凋亡的Bcl2的家族成员抵抗,如Bcl-2和Bcl-XL,它们结合并中和了促凋亡的对应分子。细胞色素c 激活caspase-3通过形成细胞色素c/Apaf-1/caspase-9复合体介导。这最终导致细胞凋亡[3]。
2、外源性途径
外源性途径是由肿瘤坏死因子受体超家族的死亡受体如Fas(CD95)和肿瘤坏死因子相关的凋亡诱导配体(TNF-related apoptosis-inducing ligand,TRAIL)启动的。配体与受体结合导致死亡受体寡聚,并通过招募适配分子FADD、caspase-8/10和抑制因子flip形成死亡诱导信号复合体(death inducing signaling complex,DISC)[6]。在 DISC 复合体上,caspase-8/10 被自动催化激活后激活下游的caspase-3[5]。 同时 DISC 复合体 上的 caspase-8/10 介 导 的Bid被切割,它将Bid转化为一种活性形式,从而触发细胞色素c 从线粒体中释放。细胞色素c 结合APAF-1,从而激活另一个启动子caspase-9,进而激活效应器caspase-3[6]。
循环系统
1、GC 可通过 ERS 或连接蛋白(junction-mediating and regulatory protein,JMY)诱导细胞凋亡和功能障碍来损伤血管内皮细胞
虽然ERS是内质网中错误折叠的蛋白质大量地积累引起,但是内质网可激活未折叠蛋白反应来缓解这种压力。在应激状态下,内质网最初试图通过未折叠蛋白反应(unfolded protein response,UPR)信号恢复内环境平衡,如果UPR 诱导的各种机制不能缓解ERS,内源性和外源性的细胞凋亡通路都可以被激活[7]。
1.1、蛋 白 激 酶 样 ER 激 酶(PKR-like endoplasmic reticulum kinase,PERK)-eIF2α - 激 活 的 转 录 因 子4(activating transcription factor 4,ATF4)-转录因子C/EBP 同源蛋白(C/EBP homologous protein,CHOP)信号通路 ERS可激活应激感受器,包括PERK、激活的转录因子6(ATF6)和肌醇需求激酶1(inositol-requiring enzyme 1,IRE1),参与细胞内稳态的调节。已知ERS 激活3 个主要信号通路,即PERK-ATF4 轴、ATF6 信号转导和 IRE1a 级联反应,对于血管内皮细胞PERK-ATF4 通路起关键作用。当感受到ERS时,PERK寡聚并磷酸化自身和翻译起始因子eIF2α,从而间接失活eIF2α 并抑制其翻译。通过这种方式,PERK 有助于减少进入内质网的蛋白质流量,以缓解内质网压力[8]。然而,强烈或持续的ERS 可能会破坏UPR 的动态平衡。随后PERK-CHOP 信号通路被激活,诱导细胞凋亡[9]。而IRE1a级联反应不仅能诱导内皮细胞凋亡,还可以导致内皮细胞自噬。IRE1α 信号的激活上调了X-box 结合蛋白的表达,且其与自噬、分化、耐药、低氧耐受和细胞存活相关,从而对内皮细胞的存活和增殖起到保护作用,避免了GC对细胞的进一步损伤[10]。
1.2、JMY 凋亡通路 最近有少数学者认为JMY 参与了内皮细胞的凋亡。JMY 被发现是一种损伤反应蛋白。GC处理的情况下,过表达JMY 可增加细胞的凋亡率,而下调JMY 可降低GC 诱导的内皮细胞的凋亡率。当GC 处理后Bax 表达增加时,JMY 基因的下调可显著降低GC 诱导的内皮细胞中Bax 的表达,而JMY 基因的过表达可增加GC 诱导的内皮细胞中Bax 的表达。因此,JMY 可通过上调凋亡蛋白Bax进一步诱导促凋亡途径[11]。
2、血管平缓肌的相关凋亡通路
Price 等[12]在其小鼠模型中证实地塞米松可能通过抑制核因子κB 的作用,诱导大鼠肺动脉高压大鼠肺动脉平滑肌细胞发生凋亡,从而发挥抗血管重塑的作用。另一些学者发现GC 通过抑制了miR-25 的表达,同时增加了活性氧(reactive oxygen species,ROS)的产生介导血管平滑肌细胞凋亡[13]。
神经系统
1、GR/盐皮质激素受体(mineralocorticoid receptor,MR)表达和激活的失衡通路
一些数据表明,至少在海马体中,这些效应是由不同的细胞内受体亚型介导的:GR 和 MR[14]。GR/MR 表达和激活的平衡被发现是维持海马神经元正常功能和结构的先决条件。GR激活过多或持续时间过长或MR激活和表达不足会抑制海马神经元的神经发生,促进细胞凋亡和树突萎缩,导致认知、情绪和神经内分泌障碍及空间记忆功能障碍[15]。GC 和血浆皮质酮对下丘脑室旁核细胞的存活与否具有相反的作用,并且GC的有害作用具有剂量和时间依赖性。大剂量GC 上调GR 的表达和活化,但不能上调MR 的表达,破坏了GR/MR 平衡,从而导致下丘脑室旁核中的细胞凋亡。相反,低剂量皮质醇或是血浆皮质酮通过上调MR的表达和激活发挥抗凋亡作用[16]。许多研究报道了GC 和信号通路之间的串连[17]。已有的应激模型说明了慢性应激或长期外源性GC 给药抑制海马脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)和 cAMP 反 应 元 件 结 合 蛋 白(cAMP responsive element binding protein,CREB)的表达,导致神经元凋亡;大剂量MP 对GR 的激活作用主要是通过上调Bax 表达、降低Bc1-2 表达和激活Bad 来实现的,而小剂量的皮质醇替代通过增加Bax 表达、灭活Bad 和降低Bax 表达来激活 MR,综上说明 GR 和 MR 通过 Akt/CREB/BDNF 通路调节海马细胞的凋亡与抗凋亡作用[16]。因此,GR 的激活可显著抑制蛋白激酶 B(protein kinase B,Akt)/CREB 的激活,降低BDNF 和Bcl2 的表达。相反,低剂量皮质醇对MR的激活增加了Akt/CREB 的激活,并增加了BDNF 和Bcl2 的表达。RU486 和SPIRO 分别拮抗MP 的促凋亡作用和血浆皮质酮的抗凋亡作用,也验证了它们的促凋亡即抗凋亡作用是通过GR和MR介导的[16]。
2、ERS途径
已有研究表面,血液中高浓度的GC 可以穿过血脑屏障,导致神经元损伤甚至死亡。当ERS 持续存在时,PERK和IRE1可以被激活。激活的PERK在自身磷酸化形成低聚体后,启动下游的ATF4-CHOP途径。过量的ERS可以导致IRE1 的自磷酸化,然后通过其磷酸激酶活性启动下游信号通路。作为凋亡因子,CHOP持续表达可通过阻断细胞周期和诱导细胞凋亡而导致机体损伤[18]。然而GC 也具有保护神经细胞的作用。GC 诱导的亮氨酸拉链多肽可防止视网膜上的光感受器细胞凋亡的作用,具有保护视网膜功能免于退化,因此是治疗退行性视网膜疾病的潜在选择[19]。
运动系统
1、股骨头坏死是临床常见病,致残率高
大剂量短期或长期小剂量GC 诱导的骨细胞及成骨细胞凋亡是可通过蛋白激酶C-β(PKCβ)-p66(shc)-c-Jun 氨基末端激酶(c-Jun N-terminal kinase,JNK)或是ERS 通路。其原理也成为激素性股骨头缺血坏死的一些生物学基础[20]。
1.1、PKCβ-p66(shc)-JNK 信号通路 GC 通过 GR 依赖机制激活促凋亡蛋白酪氨酸激酶2(proline-rich tyrosine kinase 2,PYK2)和JNK促进骨细胞的凋亡,从而对抗整合素诱导的存活,这种快速的细胞内信号向细胞外传导导致细胞脱离是由于细胞凋亡导致的[21]。Wang等[22]发现GC 可通过 PKCβ/p66(shc)信号转导产生 ROS,而 ROS 可以激活JNK;p66(shc)为一种线粒体中产生过氧化氢的放大因子,从而增加ROS。由此可以推断GC 对细胞的促凋亡作用需要 PKCβ/p66(shc)/JNK 信号级联的激活的ROS 导致骨细胞凋亡。同时在ROS 作用下,Forkhead box O(FOXO)转录因子的活性也升高,FOXO 的激活不仅抑制Akt的磷酸化以及Wnt 诱导的增殖和成骨细胞分化,因而ROS 的激活不仅促进细胞凋亡而且通过JNK介导的ROS诱导得细胞凋亡[23]。
1.2、ERS 途径 最近的研究表明,大剂量的GC 可以引起内质膜钙通道的改变,导致内质网钙离子耗竭或超载,从而扰乱内质网蛋白质的合成、折叠和修饰导致错误折叠的蛋白质增多导致 ERS。在 ERS 时,不仅 IRE1、PERK 和ATF-6 的激活通路都可以促进CHOP 的表达,而且ATF4 和ATF3 的合成增加,也促进 CHOP 的表达[24]。CHOP 的过量表达促进促凋亡基因的表达来诱导细胞凋亡,也因此可以抑制抗凋亡基因的表达来诱导细胞凋亡[25]。高水平的CHOP 激活了细胞的凋亡途径,加速骨细胞凋亡,导致了骨质疏松[24]。
2、骨髓间充质干细胞PERK-核因子E2 相关因子2(nuclear factor erythroid 2-related factor 2,Nrf2)凋亡通路
GC 也可导致骨髓间充质干细胞凋亡。GC 可激活PERK-Nrf2 通路促进ERS,最终影响骨髓间充质干细胞的增殖和凋亡。Nrf2是一种存续转录因子,被PERK磷酸化后从细胞质移动到细胞核,磷酸化后的Nrf2 可抑制蛋白质翻译,诱导氧化还原动态平衡基因,从而诱导细胞凋亡[26]。
消化系统
长期服用地塞米松——一种合成的GC 药物。GC 会导致高血糖或类固醇诱导的糖尿病。有研究表明地塞米松直接诱导胰腺β 细胞凋亡,但其分子机制尚不清楚。Suksri等[27]认为地塞米松通过与 GR 结合,诱导 TRAIL 死亡受体(DR5)和TRAIL 途径,诱导胰腺β 细胞发生凋亡。其在小鼠研究中发现GC不仅可上调TRAIL基因和蛋白表达,还能上调DR5 蛋白,但抑制诱骗受体蛋白。敲除DR5 可降低地塞米松诱导的 caspase-3 活性,caspase-8 和 caspase-9 抑制剂可保护胰腺β 细胞免受地塞米松促进诱导细胞凋亡,说明了地塞米松可能通过TRAIL 途径诱导胰腺β-细胞凋亡。而相关研究证明了糖原合成酶激酶3(glycogen synthase kinase-3,GSK3)参与了 GC 诱导的 β 细胞死亡和胰岛素分泌障碍。GSK3有两个亚型:GSK3α和GSK3β。GSK3β的下调导致了GR 在β细胞中的表达显著降低,单独敲除GSK3β可导致GC 促凋亡作用的消除,其揭示了β 细胞中GR-GSK3 串连的一些机制,并确定GSK3β 是导致GC 诱导的β 细胞死亡的主要亚型。有趣的是在小鼠实验中,哺乳高峰期的小鼠在接受地塞米松治疗后胰腺β 细胞质量增加。其被证实为GC 通过刺激细胞增殖和β 细胞新生,加剧了妊娠相关的大鼠胰腺β细胞质量的增加[28]。
生殖系统
GC可能通过激活Fas系统而诱导卵母细胞和卵巢细胞凋亡的假说。在体内和/或体外暴露于GC 后,卵母细胞的功能明显受损,并引发细胞凋亡,氧化应激增加,肿瘤坏死因子-α 和肿瘤坏死因子受体的表达增强。在体外,用RNA干扰或在体内敲除Fas可显著减轻GC对卵母细胞和卵巢壁层颗粒细胞的不利影响。然而,在体外,GC 显著下调输卵管上皮细胞肿瘤坏死因子α 的表达。因而Yuan 等[29]认为GC 对肿瘤坏死因子-α 表达的影响可能因细胞类型而异。有趣的是Zheng 等[30]在小鼠实验证实内源性GC 水平增高Fas系统激活增加。
免疫系统
多年来,人们对GC在免疫系统细胞中的凋亡作用进行了深入的研究,并已知GC 具有多种多效性。众所周知,大剂量的GC可诱导T细胞、B细胞、巨噬细胞、嗜酸性粒细胞、NK 细胞和胸腺细胞的凋亡[31]。有趣的是,GC 对中性粒细胞有相反的作用,实际上保护这些细胞免受凋亡[32]。
虽然许多已知的GC 诱导细胞凋亡的效应和具体机制已经在文献中得到了广泛的报道,但我们将在这里重点介绍一些关于凋亡的调节性T 细胞(regulatory T cells,Treg)分子作用机制。Treg 细胞对GC 诱导的体内凋亡具有一定水平的抵抗力。在Treg 细胞中,GC 诱导的Bcl-2 表达上调导致内源性凋亡途径的抑制,从而促进其存活。与其他胸腺细胞相比,Treg 细胞中原本较高的基础钙水平的进一步升高可能解释了它们对TCR 介导的信号的相对抵抗[33]。此外,生理性的GC 刺激表达的巨噬细胞移动抑制因子逆转GC 的免疫抑制作用,部分是通过减少激活诱导的细胞凋亡和维持炎症信号[34]。
小结与展望
GC 可通过外源性途径、内源性途径及ERS 等途径诱导细胞凋亡。临床使用GC不可避免地出现相关不良反应,但是作为临床医生充分认识GC诱导凋亡机制过程,有利于指导临床使用GC。虽然现有研究已经说明GC 具有诱导细胞凋亡的作用,但其具体机制仍旧不清楚。因此,我们仍需要努力探索研究为研制新的不良反应更少的药物提供新的理论依据。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
按摩与康复医学(2022年19期)2022-09-27
农业工程学报(2022年12期)2022-09-09
中国现代医生(2022年19期)2022-08-25
安徽医科大学学报(2021年4期)2021-04-09
科学大观园(2021年1期)2021-01-11
心理学报(2020年7期)2020-07-13
科学大观园(2019年12期)2019-09-10
课程教育研究·学法教法研究(2016年4期)2016-04-19
现代养生·下半月(2015年6期)2015-09-07
