
闽南地区21例罕见地中海贫血基因突变分析
2022-10-14 12:50蓝惠华潘燕如张梦情黄榕张玲
国际医药卫生导报 2022年19期
蓝惠华 潘燕如 张梦情 黄榕 张玲
陆军第七十三集团军医院厦门大学附属成功医院检验科,厦门 361003
地中海贫血(又称地贫)是全球最常见的遗传性疾病之一,由α-珠蛋白和β-珠蛋白基因组突变引起。地贫主要分布在地中海、非洲、中东地带、印度和东南亚的沿海地区[1]。在中国,地贫主要散布在中国南方,特别是广西、广东和海南,福建区域也有一定的地贫基因的携带量,尤其是毗邻广东的闽南地区[2-4]。重型地贫患者往往胎死子宫内或婴儿早期死亡,而中型地贫患者则往往表现为终身贫血,且必须长期输血和祛铁治疗,给家人和社区带来了很大压力[5]。所以,地贫携带者的检测和产前诊断,对减少地贫高发区域内的地贫病死率必不可少。目前,针对我国南方人群常见类型α、β地贫的检测,市面上通用的几种地贫基因检测试剂盒基本上都能涵盖,不过,由于地贫基因的分子基础相对复杂、突变种类众多,在临床工作中也会碰到一些血液学表型与基因突变检测结果明显不相符,可能存在罕见突变基因的例子[6]。我们收集了2017年11月至2022年5月来厦门大学附属成功医院就诊的38例临床血液学检测异常但常规地贫基因突变检测结果阴性,可能含有罕见地贫基因突变的样本38份,使用了跨越断裂点聚合酶链反应(GAP-PCR)和电泳、基因测序检查方法对其进行了检测评价,用以进一步明确诊断并逐步扩充本地区的地贫基因突变谱。
资料与方法
1、一般资料
收集了2017年11月至2022年5月来厦门大学附属成功医院就诊的临床血液学检测异常但常规地贫基因突变检测结果阴性的患者38例,其中男16例,年龄5~47岁;女22例,年龄15~56岁。38例样本分别来自福建闽南地区的9个不同地方,包括厦门思明区、厦门同安区、漳州龙海区、漳州芗城区、漳州云霄、漳州诏安、泉州晋江、泉州安溪、泉州石狮。样本采用EDT A-2K抗凝的真空管静脉血2 ml,在4℃环境下邮寄送至广东凯普生物有限公司进行测序分析。患者已在厦门大学附属成功医院进行了血液学分析、血红蛋白电泳分析和地贫常规基因突变的检测。上述样本的测序检测与广东凯普生物有限公司合作完成。全部的检测都通过受试者的知情同意,并签订了知情同意书。罕见α-地贫的纳入标准[7]:血红蛋白H(Hb H)病表型而基因型仅为标准型的患者;生育过Hb H病患儿而基因型正常的父母;非缺铁性的小细胞低色素贫血,基因型正常的患者。罕见β-地贫的纳入标准[7]:血红蛋白(Hb)电泳HbA2>3.5%,且呈小细胞低色素贫血而基因型却是正常的患者;重型地贫表型而基因型仅为携带者的患者。
2、检测方法
血液学常规分析采用深圳迈瑞CAL8000自动化血细胞分析流水线及其配套试剂进行检测;血红蛋白电泳运用HYDRASYS 2全自动电泳仪进行分析;运用Veriti Dx96聚合酶链反应(PCR)扩增仪及HHM-2医用核酸分子快速杂交仪,对常见的3种α-地贫缺失基因(--SEA、-α3.7和-α4.2)、3种α-地贫突变基因(CS、QS、WS)及IVS-II-654(C-T)、-28(A-G)、-29(A-G)、CD14∕15(+G)、CD17(A-T)、CD27∕28(+C)、βE(G-A)、CD41∕42(-TTCT)、CD43(G-T)、CD71∕72(+A)、IVS-I-1(G-T,G-A)、IVS-I-5(G-C)、Cap(-AAAC)、Int(T-G)、CD31(-C)等15个β-地贫基因突变位点通过GAP-PCR方法、PCR扩增和导流杂交的原理实现[8]。全血DNA提纯、PCR扩增和PCR产物杂交均采用由广东凯普生公司提供的试剂盒,严格规定遵照操作程序执行各项检测。
α及β地贫的测序分析利用Sanger双去氧链终止法完成,先利用外周血中DNA提取试剂盒抽取基因组DNA,再利用GAP-PCR技术测定缺失型α及β地中海基因,对PCR的扩增物质采用琼脂糖凝胶电泳,确定得到目的片段后,再将扩增产物与之对应的引物,送到广州凯普公司进行测序技术分析,在完成测序技术后,通过与http:∕∕blast.ncbi.nlm.nih.Gov∕对比,分析测序图纸,以判断是否有新突变位点的存在,扩增引物和反应条件参照文献[6]。
结 果
38例可疑为罕见地贫的样本中检出21例为稀有类型地贫,当中包含了6种罕见的α珠蛋白基因突变,分别为8例泰国型缺失(--THAI)、2例中国香港型缺失(HKαα)、2例菲律宾型(--FIL)、1例CD 30(-GAG)、1例CD 122 CAC>CAG,WSM和1例αα∕αααanti4.2;其中2例--FIL源于一对母子。6种罕见β珠蛋白基因突变,分别为2例IVS I-129 A>G杂合突变、1例CD56 GGC>GAC杂合突变、1例CD54 GTT>GTC,Val>Val杂合复合Terminal CD+32 A>C杂合突变、1例113A>G(Poly A(A>G)AATAAA>AATAAG)杂合突变,1例越南型(Vietnamese)遗传性胎儿血红蛋白持续症(hereditary persistence of fetal hemoglobin,HPFH)缺失型β地贫,其中CD54 GTT>GTC杂合突变以往未见报道,其测序图见图1。21例罕见地贫的基因突变详见表1。
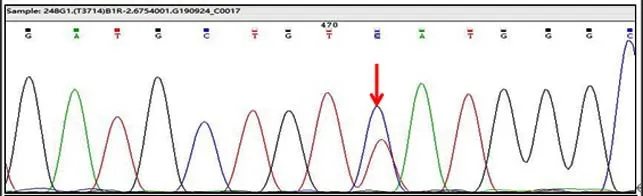
图1 CD54 GTT>GTC杂合突变测序结果
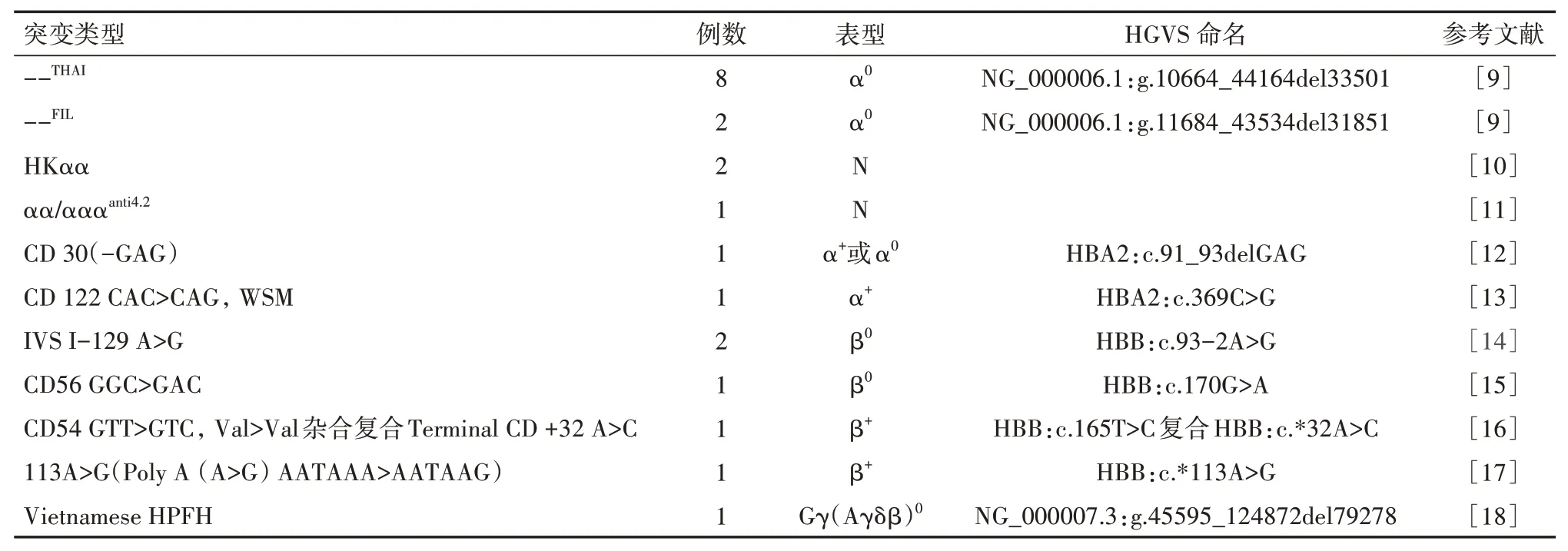
表1 21例罕见地贫的基因突变类型
讨 论
本研究通过对38例疑似罕见地贫患者的血液样本进行测序分析,共发现21例罕见型地贫,阳性率高达55.26%,其中α-地贫15例,α地贫以--THAI最常见(占53.33%),其次是--FIL和HKαα。α地贫在整个东南亚很常见,最常见的原因是16号染色体短臂上α珠蛋白基因簇的不同DNA片段缺失,该基因复合体包括2个α基因(α2和α1)、1个胚胎基因(ζ2)、几个假基因(ψα1、ψα2、ψζ1)和1个功能未知的基因(θ1),顺序为5’-ζ2-ψζ1-ψα1-ψα2-α2-α1-θ1-3’[19]。在整个热带和亚热带地区,2个α基因(αα)中的1个缺失(-α)是常见的,而涉及2个α基因(--)的缺失最常见于东南亚和地中海盆地,即Hb H病(--∕-α)和Hb Barts胎儿水肿综合征(--∕--)的高发地,在东南亚,以--SEA缺失型最常见,它是约20 kb长度的α基因簇的缺失,即α1和α2基因完全缺失,仅保留ζ2和ψζ1基因不变[19]。然而在泰国和菲律宾地贫患者中首次发现其中一条染色体上的ζ-α复合体完全缺失,因而分别称之为--THAI和--FIL,由于这些缺失延伸到5’端的ζ基因之外和3’端的α基因之外,它们位于θ1基因之外的一个小序列DNA区域附近,该区域被命名为a-珠蛋白3'高变区(3'HVR),因此通过珠蛋白复合物的常规限制性图谱,不能在杂合子(--∕αα)中明确识别它们,因此在遗传咨询和产前检测期间可能会遗漏这些缺失[9]。--THAI和--FIL这2个缺失被定位到5’断点的ζ2珠蛋白基因上游约4 kb的区域,--FIL缺失30~34 kb,--THAI缺失长度为34~38 kb,与3'HVR不同的是,用于识别5'断点的探针可识别对2种突变中的每一种都具有特异性的独特片段,并可用于常规产前检测[9]。与--SEA相比,--THAI和--FIL突变在Hb H疾病和Hb Bart胎儿水肿综合征受试者中相对少见或表达不足,需要进行更广泛的研究,以确定其在这些人群中的真实频率。--THAI或--FIL突变的纯合子中可以产生由ε4或γ4制成的血红蛋白,这两者都不可能支持发育中胚胎和胎儿的氧气需求,因此,受影响父母的后代(--∕αα)很可能出现高频率的早期自然流产,这些突变体的复合杂合子和常见的东南亚决定基因(--FIL∕--SEA或--THAI∕--SEA)也会患有Hb-Bart胎儿水肿综合征,并且在早期胚胎和胎儿生命中也可能受到严重损害,因为只有一个功能性ζ-珠蛋白基因,这也可能导致早期流产。在东南亚人群中的“高危”夫妇中鉴定这些突变体尤为重要,尤其是采用常规方法无法鉴定识别的--FIL∕αα和--THAI∕αα携带者。本研究发现2例HKαα。HKαα异常基因型因2005年在中国香港首次报道检出而命名,是一种极少见的在α-珠蛋白基因簇同源区域错位与减数分裂过程中的发生的不平衡交叉连接,它同时含有-α 3.7缺失及αααanti4.2基因等位基因的交叉连接片段,HKαα的基因组成成分有1个α2基因和1个由α2和α1部分片段组成的融合基因(即右缺失片段)[10,20]。HKαα等位基因既不包含缺失,也不包含三倍体,因此这种等位基因的携带者不太可能受到任何有害影响,其血液学和临床表现都是正常的。然而,这种重排在个体中的存在对于基于PCR的α地贫分子诊断具有重要意义,临床采用的常规检测方法只能检测-α3.7缺失,不能检测到αααanti4.2,因而常常误诊为-α 3.7∕--,而导致产前咨询时误导临床给出错误的建议。
本研究检出1例αα∕αααanti4.2基因型。根据αααanti4.2基因的分子生物学特性,这个基因的载体表达的α链比野生型个体多,这将导致比野生型个体更多的α链合成,从而导致α链与β链的比例失衡[10]。当个体仅携带αααanti4.2基因时,α链与β链的比率略有不平衡,携带者不会遇到严重问题,然而,当它们同时携带β地贫等位基因时,合成α链与β链的比率进一步失衡,导致不同程度的贫血。大多数临床机构使用的常规检测试剂的检测范围不包括αααanti3.7、αααanti4.2和HKαα基因,因此,很难常规检测这些变化,由于检测方法的复杂性,以往的研究没有充分研究这些变异在人群中的携带情况,大多数检测机构也没有对其予以重视。
本研究检出1例CD 30(-GAG)突变,该突变为a2基因外显子1的密码子30缺失,即α2基因编码序列的91-93号碱基GAG缺失,然而,这并不影响该区域的剪接位点一致性序列,因为外显子的最后2个核苷酸(AG)和中间序列中的前6个核苷酸保持完整,这种框内缺失导致少1个氨基酸的α珠蛋白链,截短的蛋白质高度不稳定,因新合成的珠蛋白链分子疏水性增加,会被蛋白质水解迅速破坏,除非它们被并入Hb四聚体中[11]。CD 30(-GAG)为致病性突变,临床表型为α⁺或α0。本研究检出1例Hb Westmead,这是由HBA2密码子122(CAC>CAG)突变引起的a链变体,是一种沉默的非缺失的a+-地贫,其a-珠蛋白链缺失程度比缺失型a+-地贫轻[12]。Hb Westmead在中国南方很常见。据报道,在广西省,Hb Westmead的等位基因频率为1.55%,其次是Hb CS(1.21%)和Hb QS(0.36%)[21]。然而,这种变体不如其他常见的非缺失型α-地贫如Hb-CS和Hb-QS那样广为人知,原因可能是Hb Westmead仅与轻度α-地中海表型相关,再则在常规电泳或高效液相色谱(HPLC)上无法检测到这种变体,因而常常被忽略。
β-地贫是世界上最常见的单基因疾病之一,其特征是血红蛋白的β-珠蛋白链合成减少或缺失,严重的β-地贫会导致严重的贫血,需终生输血。突变类型与临床表现高度相关,在世界不同地区已经描述了200多个导致β-地贫的突变[22]。尽管每个人群中发现的这些突变数量相对较少。在中国人群中,常见的几种突变占所有病例的90%以上,而罕见突变对于受影响家庭的产前诊断也很重要,对于深入了解珠蛋白基因调控的潜在过程以及基因型-表型关系的研究也不可或缺。本研究检出罕见β-地贫6例。
IVS I-129 A>G突变(HBB:c.93-2A>G)是β基因编码区序列的271号碱基G突变为T,对应的编码氨基酸由谷氨酸改变为终止密码子,为致病性突变,临床表型为β0,在德国首次报道[23]。本研究发现2例该突变,且2例非亲缘关系。CD56 GGC>GAC突变,即Hb J-Bankok,由β-珠蛋白链56位的甘氨酸由天冬氨酸取代而定义,这种突变似乎产生了一种带有额外负电荷的β亚单位变体,因为β56(D7)Asp链有更多机会与带正电荷的α链结合,与正常β链竞争,从而允许在杂合子状态下形成比正常Hb A更多的Hb J-Bankok,这种Hb变体显示出快速的电泳迁移率[14]。Hb J-Bankok无临床症状。本研究检出1例,该突变者,血红蛋白电泳检出异常血红蛋白所占比例高达51.3%。Terminal CD+32 A>C突变在福建及广东广州均有报道为β基因3'端非编码区域的32号碱基A突变为C[16,24]。该突变使β珠蛋白基因型转录效率降低,mRNA稳定性低于正常水平,影响正常的β珠蛋白链的合成,为致病性突变,临床表型为β+,由于病例数的局限性,该突变的致病性有待获取更多病例后再进一步研究确认。本研究中检出的Terminal CD+32 A>C突变患者同时检出一种以往未见报道的突变类型,即HBB:c.165T>C(CD54GTT>GTC,Val>Val)突变,该突变是β基因编码区序列的165号碱基T突变为C(图1),对应的54位编码氨基酸为缬氨酸未发生改变,不影响β珠蛋白的合成,理论上不引起相关的临床症状,该突变对人体是否有其他影响及其详细机制有待进一步研究确认。
HBB:c.*113A>G突变位于HBB基因转录的起始位点上游启动子区域,为β基因3'端非编码区域的113号碱基A突变为G,为致病性突变,临床表型为β+[25]。本次检测发现该突变携带者血常规红细胞相关参数基本正常的,仅Hb电泳发现HbA2略高于正常(3.8%),无明显的临床症状,与陈扬等[25]研究推测HBB:c.*113A>G杂合突变致病类型为静止型是相符的。本次检出1例Vietnamese HPFH,它是一种与HPFH表型相关的30 kb珠蛋白基因簇缺失,越南GγAγ HPFH缺失在δ-珠蛋白基因下游有一个独特的5’断点3.5 kb,3'断裂点位于HPFH-3断裂点上游约8 kb处,位于HPFH-4的3'断点区域内[18]。进一步的证据表明,3'断点区域包含功能上重要的序列,这些序列与γ-珠蛋白基因并列是胎儿血红蛋白水平升高的一个重要因素[26]。
地贫重在预防和控制,本研究主要是收集闽南地区部分可疑地贫但常规试剂盒检测不出的样本,应用GAP-PCR和电泳、基因测序进行分析,检出了几种罕见的地贫基因突变,并介绍了这些罕见地贫基因的基本情况,丰富了闽南地区地贫相关基因突变谱,为今后闽南地区可疑地贫患者的诊断提供检测思路,不能局限于常规试剂盒的检测,对于可疑样本应采用更全面的方法进行检测,以防漏诊。本研究遗憾之处是未对各罕见地贫携带者进行家族人群进行地贫检测确认,对携带罕见地贫基因的人群进行家族可疑人群地贫检测追踪,有望挖掘出更多罕见类型地贫,这也是我们今后工作需要加强的地方。罕见地贫基因型虽在人群中占比不高,但依然不能忽视针对罕见地贫突变的研究,相信未来随着检测技术的发展,更多的突变类型将不断被发现,而某些罕见类型也将成为不罕见。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
作物学报(2022年12期)2022-10-14
中国现代医生(2022年21期)2022-08-22
安徽农学通报(2022年6期)2022-04-07
瞭望东方周刊(2021年6期)2021-03-30
瞭望东方周刊(2021年6期)2021-03-30
三农资讯半月报(2020年2期)2020-03-09
新课程·下旬(2018年8期)2018-11-10
中学生理科应试(2017年6期)2017-09-27
中学生理科应试(2017年2期)2017-04-01
中学生理科应试(2016年4期)2016-11-19
