
四个CyB5Q[5]-Ca配合物的晶体结构和在不同阴离子环境下的不同配位状态
2022-10-10 05:43:00邓德朋陈明华刘元东钱丙飞夏位敏易君明
无机化学学报 2022年10期
邓德朋 陈明华 刘元东 钱丙飞 夏位敏 易君明
(兴义民族师范学院生物与化学学院,兴义562400)
瓜环(cucurbit[n]uril,Q[n],n=5~8、10、13~15)是一类甘脲通过亚甲基连接而成的环状化合物,是超分子化学领域的一类研究较活跃的化合物。瓜环作为主体可以其带负电性端口的羰基氧原子与大多数金属离子形成各种类型的瓜环-金属配合物[1-3],也可利用其电中性的空腔,识别、自组装形成主-客体聚合物[4-5],还能利用电正性的外壁,与其它客体、瓜环形成主-客体外壁和主体-主体外壁-端口聚集体。这些超分子聚集体形成的驱动力除了金属配位键外,大多是通过分子间的非共价键相互作用,如氢键、偶极-偶极作用、C—H…π作用、π…π堆砌和疏水作用等[6-8]。
以瓜环为主体的超分子化学已经显示出广泛的应用前景。目前,瓜环化学的研究内容包括:(1)合成新型瓜环,增加瓜环的种类,目的是改变瓜环的水溶性,改变瓜环的不可修饰性等[9-13];(2)设计、合成不同的瓜环基主客体超分子聚集体,研究它们对光、电、磁、pH和热等外界条件的响应,探索它们作为新型材料的潜在应用[14-18]。
近年来,基于瓜环化学研究,陶朱教授团队针对瓜环基金属配合物,提出了“有机结构导向剂(organic structure-directing agent,OSDA)”概念[19],即瓜环在形成超分子聚集体的过程中,某些物质(客体)的存在可以诱导瓜环-瓜环、瓜环-客体和瓜环-金属间形成结构特异的超分子有机骨架(supramolecular organic frameworks,SOFs),这类SOFs作为一种新型多孔材料,在气体的储存和分离、探针、药物传输和催化等领域有广泛的应用[20]。
环丁基取代瓜环(cyclobutanocurbit[n]uril,CyBnQ[n],n=5~8)作为一类新型瓜环,首次被澳洲新南威尔士大学Day团队于2017年报道[21]。我们实验室于2018~2020年间合成了该类物质,它们的结构第一次被X射线单晶衍射技术确定(图1a),其与经典瓜环的区别在于前者的空腔内有更多的负电荷分布(图1b)[22]。
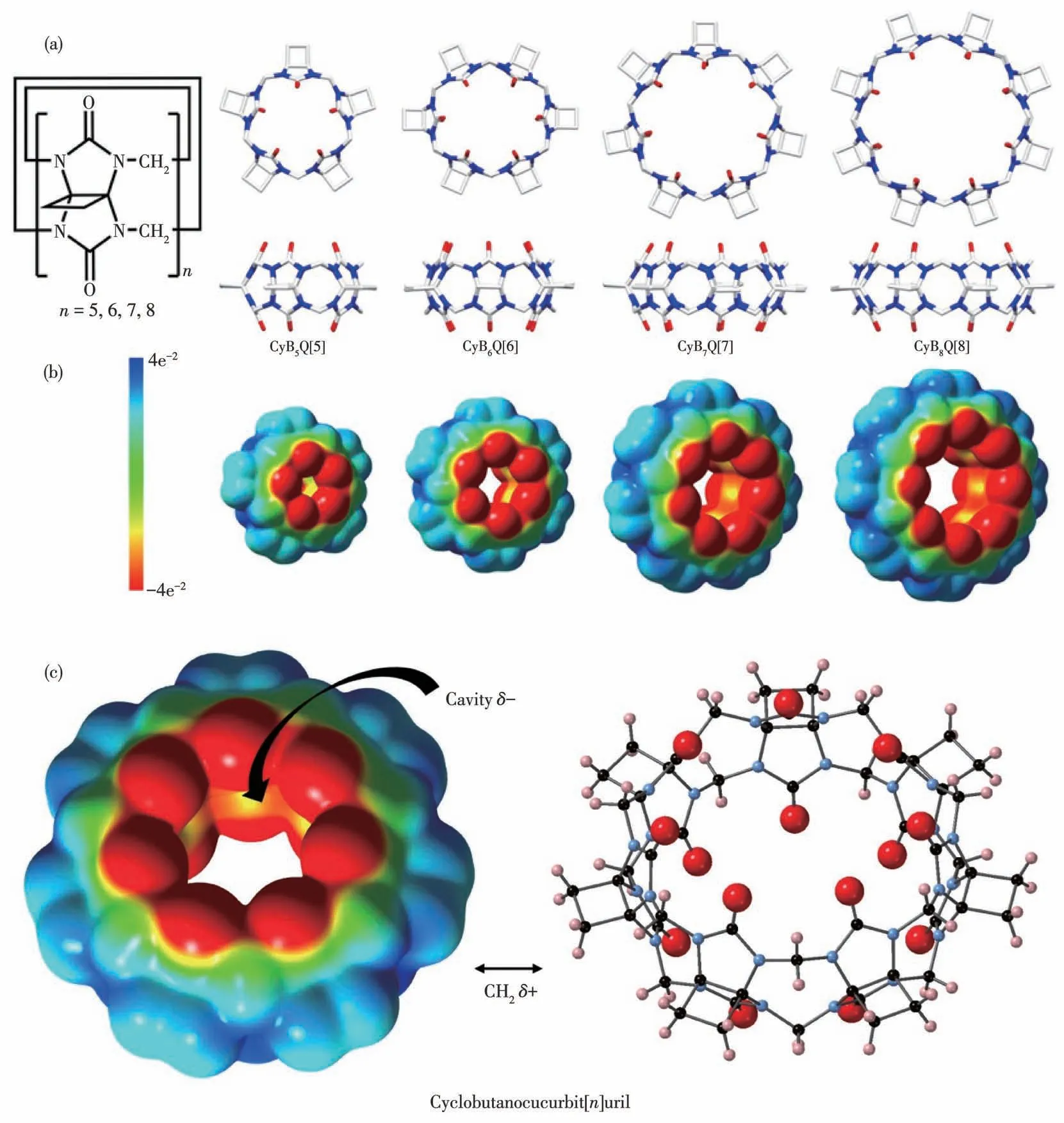
图1 (a)CyBnQ[n]的结构简图和晶体结构[22];(b)CyBnQ[n]的电荷密度分布图(EPS)[22];(c)CyBnQ[n]的电负性内壁和电正性外壁示意图Fig.1(a)Structural brief diagrams and crystal structures of CyBnQ[n][22];(b)Electrostatic potential maps of CyBnQ[n][22];(c)Electronegative cavity and electropositive external walls of CyBnQ[n]
在CyBnQ[n]的合成、分离和纯化过程中,我们发现,无论是合成反应中析出的瓜环,还是硅胶柱分离、纯化得到的瓜环,即使没有加入任何钙盐,CyB5Q[5]组分总是被钙离子配位。这就意味着合成、分离环境中的微量钙离子,也可被CyB5Q[5]捕捉到,并形成CyB5Q[5]-Ca配合物。为了证实这个推断,将分离、纯化后的CyB5Q[5]经X射线光电子能谱(XPS)测试,结果显示,化合物中确实有钙元素存在(图S1,Supporting information)。文献中[23]碱土金属钙与Q[5]的配位常数也支持了这个结论,即钙离子与Q[5]有较大的亲和能力。
为了进一步了解钙离子在不同的介质环境下与CyB5Q[5]生成配合物的情况,我们使用了不同的阴离子与CyB5Q[5]作用,得到了不同配位数的CyB5Q[5]-Ca配合物,本文介绍它们的配位情况。本文也是第一次报道新型瓜环家族成员CyB5Q[5]的钙离子配合物晶体结构。
1 实验部分
1.1 主要试剂和仪器
HClO4(40%)、ZnCl2(无水)、HCl(浓)均为市售分析纯试剂,未经处理直接使用。元素分析(C、H和N)采用Elementar Vario EL cube型元素分析仪(德国)进行。热重分析(TGA)采用Netzsch STA2500型(德国)进行,载气为N2,升温速率为10℃·min-1,温度范围:25~800℃。粉末X射线衍射(PXRD)采用日本理学SmartLab进行,辐射源及其波长:CuKα(0.154 056 nm),电压:40 kV,电流:40 mA,扫描范围:5°~90°。
1.2 单晶的制备
1.2.1 化合物1的制备
CyB5Q[5]的合成参照文献[21]。将合成得到的混合CyBnQ[n](n=5~8)蒸除盐酸,残渣加水溶解,滤掉不溶物后,滤液再蒸除水,重复3次,去除大部分盐酸。残余物烘干后再加蒸馏水溶解,溶液过滤,静置2周后析出晶体为CyB5Q[5]。
1.2.2 化合物2的制备将0.250 g前述CyB5Q[5]加入100 mL蒸馏水中,再加浓盐酸1 mL,加热溶解后过滤,冷却后的滤液备用。
取5 mL备用液,加入0.002 g无水ZnCl2,超声振荡溶解后,过滤,清液静置3周后,得到单晶。
1.2.3 化合物3的制备取2的备用液5 mL,加入5滴HClO4,与2的溶液等量混合,过滤,室温静置3周后得到单晶。
1.2.4 化合物4的制备取2的备用液5 mL,加入5滴HClO4,过滤,清液静置3周后,得到单晶。
1.3 单晶结构的测定
上述单晶在Rigaku Oxford Diffraction Supernova Dual Source衍射仪(Oxford Diffraction Ltd.,Abingdon,England)上收集数据,使用石墨单色化的MoKα射线(λ=0.071 073 nm),扫描方式为ω-scan模式,采用了洛伦兹极化法和吸收校正法。分别使用SHELXT-14和SHELXL-14程序包进行了基于F2的结构解析和全矩阵最小二乘细化,全部数据经Lp因子和多重扫描吸收校正,使用Olex2软件和SHELXS程序解出晶体结构,用SHELXL程序对全部非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法精修至收敛。由理论加氢法给出氢原子。晶体学数据和结构修正参数见表1。
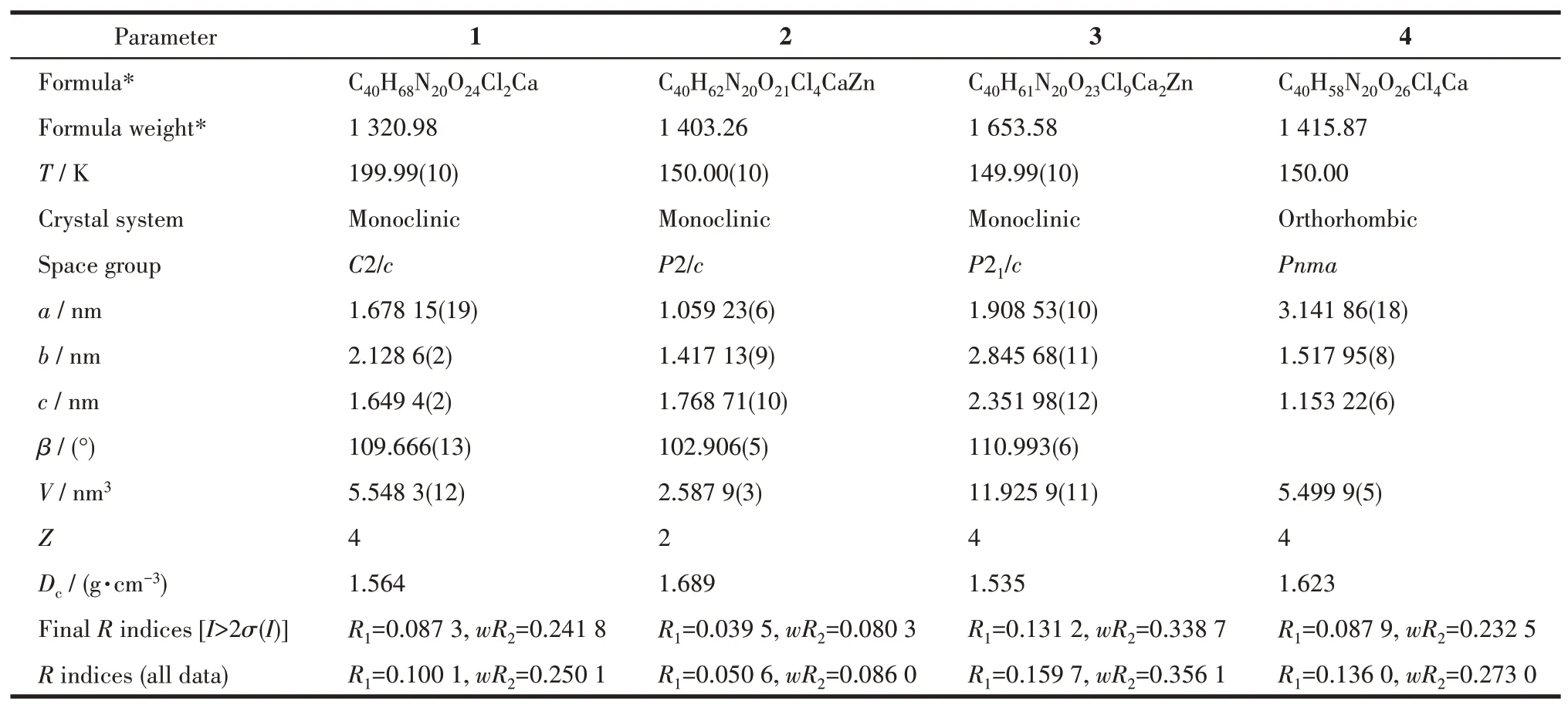
表1 化合物1~4的晶体学数据Table 1 Crystallographic data for compounds 1-4
CCDC:2126404,1;2126405,2;2126406,3;2126407,4。
2 结果与讨论
2.1 化合物1的晶体结构
由图2可见,2个钙离子Ca1和Ca1#3分别与同一个CyB5Q[5]的两端羰基氧原子O3、O4和O3#2、O4#3配位,再分别与水分子的氧原子O1W#1、O1W和O1W#2、O1W#3配位后,同时与另2个CyB5Q[5]端口的O3#1、O4#1和O3#2、O4#2形成2个六配位的“分子胶囊”(图2b),O3—Ca1、O3#2—Ca1#3、O4#2—Ca1#3、O4—Ca1的 距 离 为0.226 9 nm,O1W#1—Ca1、O1W#2—Ca1#3、O1W—Ca1、O1W#3—Ca1#3的距离为0.232 3 nm,即钙离子与不同类型的氧原子在CyB5Q[5]两个端口形成正八面体的配位构型(图2b)。
图3为化合物1的1D分子链间的氢键作用(3个CyB5Q[5]间的氢键作用类型一样,为清晰显示,删除了重复部分)。2个CyB5Q[5]间除了通过图2b所示的配位键连接外,CyB5Q[5]的氧原子(O1#1、O2#1、O5#5和O1#1、O2#2、O5)还分别以配位水分子的O1W、O1W#3通过多个氢键连接,并使2个CyB5Q[5]端口完全封闭,这些氢键的细节及键长和键角分别见图3和表2(1D分子链中的氢键参数值在重复结构中完全相等,表2中的原子没有标出对称操作符)。
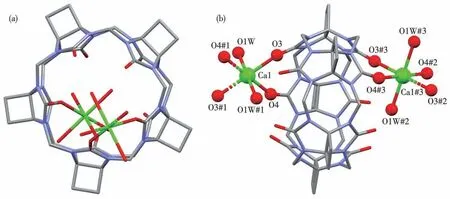
图2 化合物1的晶体结构图:(a)俯视;(b)侧视Fig.2 Crystal structure diagram of compound 1:(a)top view;(b)side view
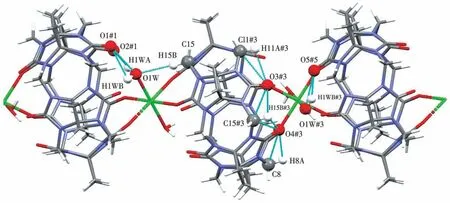
图3 化合物1的1D分子链中的氢键Fig.3 Hydrogen bond in the 1D molecular chain of compound 1
图4a为化合物1的3D堆积图。可以看出,有大量的氯离子和水分子填充在1D分子链的空隙中,形成孔洞。图4b为2D堆积中非共价键相互作用(省略了水分子和氯离子)。图4c为2D堆积中非共价键相互作用的细节,它们为2个瓜环腰上的环丁烷环碳原子和氢原子形成相同的氢键(表2)和一个偶极-偶极作用(H18A…H18A 0.233 3 nm)。
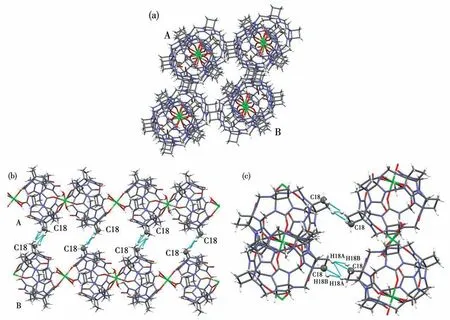
图4 (a)化合物1的3D堆积(A、B表示2个1D分子链在堆积中的位置);(b)2D堆积中非共价键相互作用;(c)A和B分子链间的非共价键相互作用细节Fig.4(a)Two-dimensional stacking of compound 1(A,B indicates the position of two 1D molecular chains in the packing);(b)Non-covalent bond interactions in the 2D stacking;(c)Details of non-covalent bond interactions between A and B molecules chains
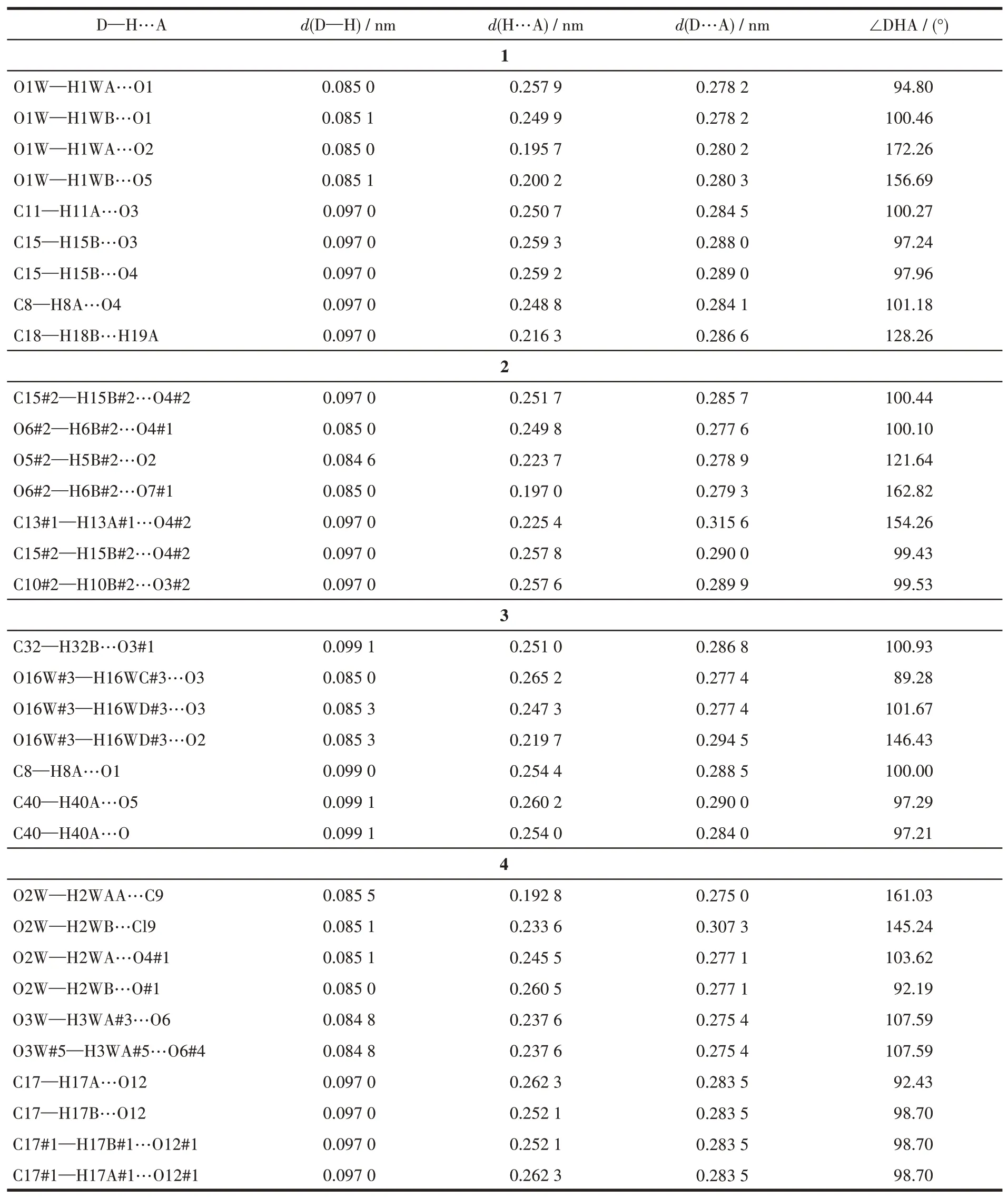
表2 化合物1~4中的氢键Table 2 Hydrogen bonds of compounds in 1-4
2.2 化合物2的晶体结构
图5为化合物2的基本结构单元图,其中Ca1、Ca1#3除与1个CyB5Q[5]的2个端口的氧原子(O3、O4和O3#2、O4#2)分别直接配位外,还分别与水分子中的氧原子(O5、O6、O5#1、O6#1、O5#2、O6#2、O5#3、O6#3)配位,同时还分别与另2个CyB5Q[5]的O3#1、O4#1和O3#3、O4#3配位,形成八配位的配位构型(图5b),这些配位键包括O3—Ca1(0.252 0 nm)、O4—Ca1(0.248 3 nm)、O5—Ca1(0.238 4 nm)、O6—Ca1(0.240 1 nm)。通过这些配位键,CyB5Q[5]进一步延伸为1D分子链。
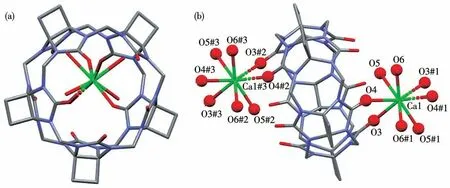
图5 化合物2的晶体结构图:(a)俯视;(b)侧视Fig.5 Crystal structure diagram of compound 2:(a)top view;(b)side view
图6为化合物2的1D分子链中的非共价键作用。2个CyB5Q[5]分子间除了配位键外,还以配位水分子的氧原子(O5、O6)与未配位的端口氧原子(O1、O2、O7)之间的偶极-偶极作用,将CyB5Q[5]两端封口。这些偶极-偶极作用包括O6#4…O7#4(0.279 3 nm)、O5#4…O2#1(0.278 9 nm)、O5#4…O1#1(0.269 0 nm)。同时,多个氢键将瓜环2个端口封闭(图6b和6c),这些氢键的键长及键角见表2。
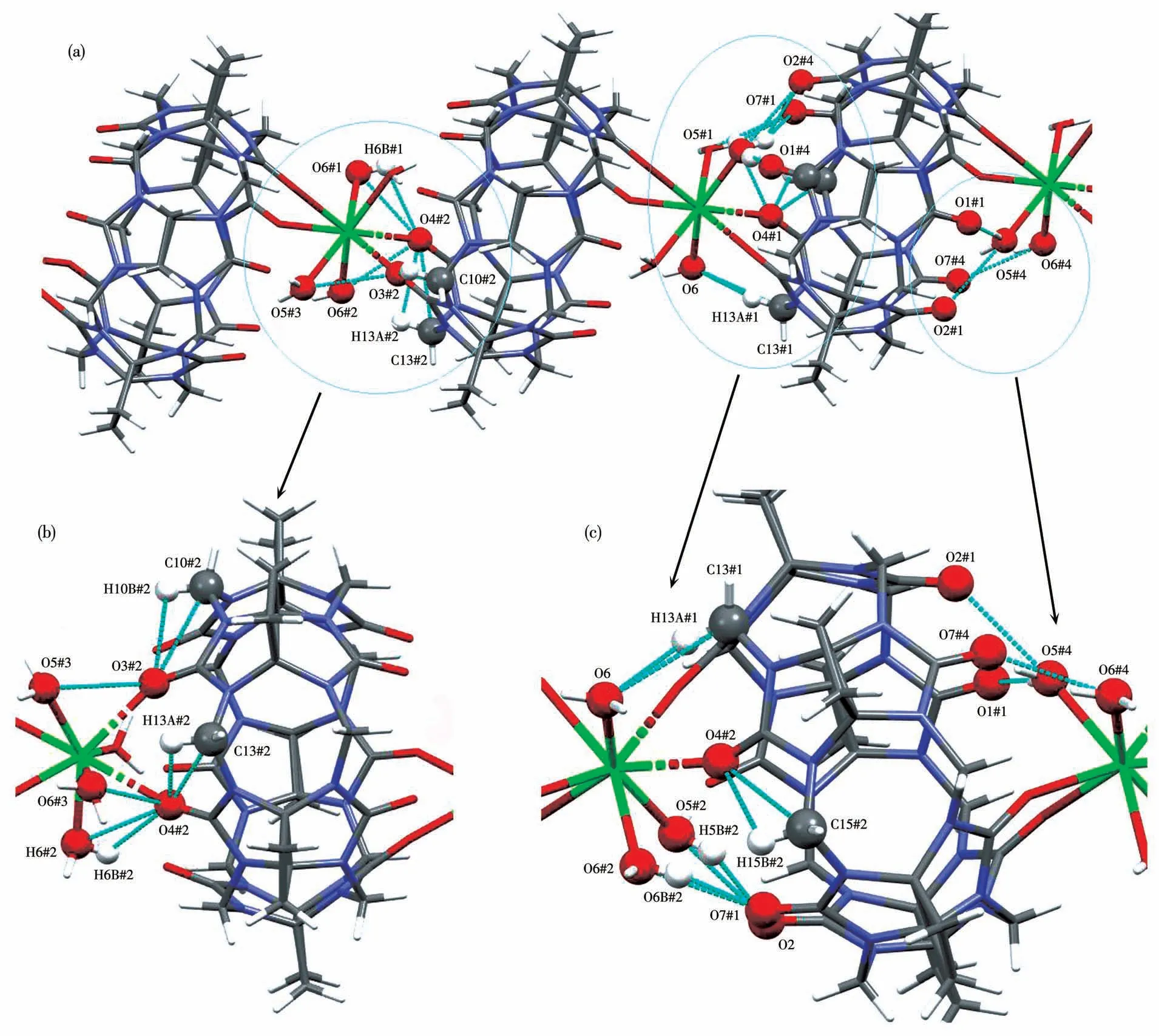
图6 化合物2的1D分子链:(a)非共价键相互作用;(b、c)非共价键相互作用细节Fig.6 One-dimensional molecular chain of compound 2:(a)non-covalent bond interactions;(b,c)details of non-covalent bond interactions
图7为化合物2的3D堆积图(图7a)和2D分子链中的非共价键相互作用(图7b)。在2个一维分子链A、B间,通过[ZnCl4]2-中的4个氯离子,分别形成偶极-偶极作用:H3B…Cl2(0.274 7 nm)、H19B…Cl1(0.293 7 nm)、H16A…Cl1(0.219 1 nm),将2个1D分子链连接为2D分子链。一般来说[ZnCl4]2-不稳定,但在本实验的条件下,能作为1D分子链间的连接介质而稳定存在。
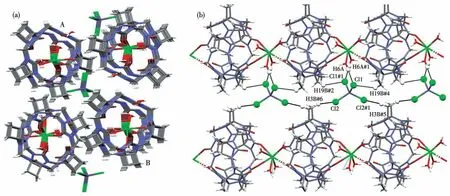
图7 化合物2的(a)沿a轴观察的3D堆积和(b)2D堆积中的非共价键相互作用Fig.7(a)Three-dimensional stacking viewed along the a-axis and(b)non-covalent bond interactions in 2D stacking for compound 2
2.3 化合物3的晶体结构
图8为化合物3的基本结构单元。2个Ca分别与1个CyB5Q[5]两端的氧原子(O11、O12、O16、O17)配位,同时与另2个CyB5Q[5]端口氧原子(O6、O10、O1#4、O5#4)配位,再分别与水分子(O1W、O2W、O3W和O15W、O16W、O17W)配位,形成2个七配位不规则九面体的配位构型(图8b)。这些配位键包括Ca1—O11(0.230 7 nm)、Ca1—O12(0.243 6 nm)、Ca1—O6(0.247 9 nm)、Ca1—O10(0.230 2 nm)、Ca1—O1W(0.233 8 nm)、Ca1—O2W(0.239 2 nm)、Ca1—O3W(0.238 9 nm)、Ca2—O1#4(0.236 6 nm)、Ca2—O5#4(0.237 0 nm)、Ca2—O16(0.231 1 nm)、Ca2—O15W(0.232 4 nm)、Ca2—O16W(0.232 1 nm)、Ca2—O17W(0.261 2 nm)。
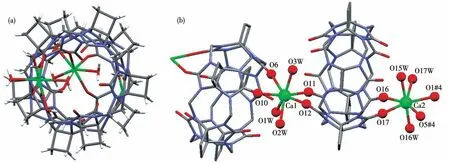
图8 化合物3的结构单元:(a)俯视;(b)侧视Fig.8 Structural unit of compound 3:(a)top view;(b)side view
图9为化合物3的1D分子链中的非共价键相互作用,包括偶极-偶极作用和氢键。偶极-偶极作用包括O3W…O14(0.290 9 nm)、OW…O15(0.288 8 nm)、O15W#3…O18#3(0.278 8 nm)、O15W#3…O19W#3(0.284 6 nm)、O15W#3…O20#3(0.205 3 nm)、O17W#3…O20#3(0.262 3 nm)、Ca2#3…H47A#3(0.318 2 nm)。氢键的键长和键角见表2。
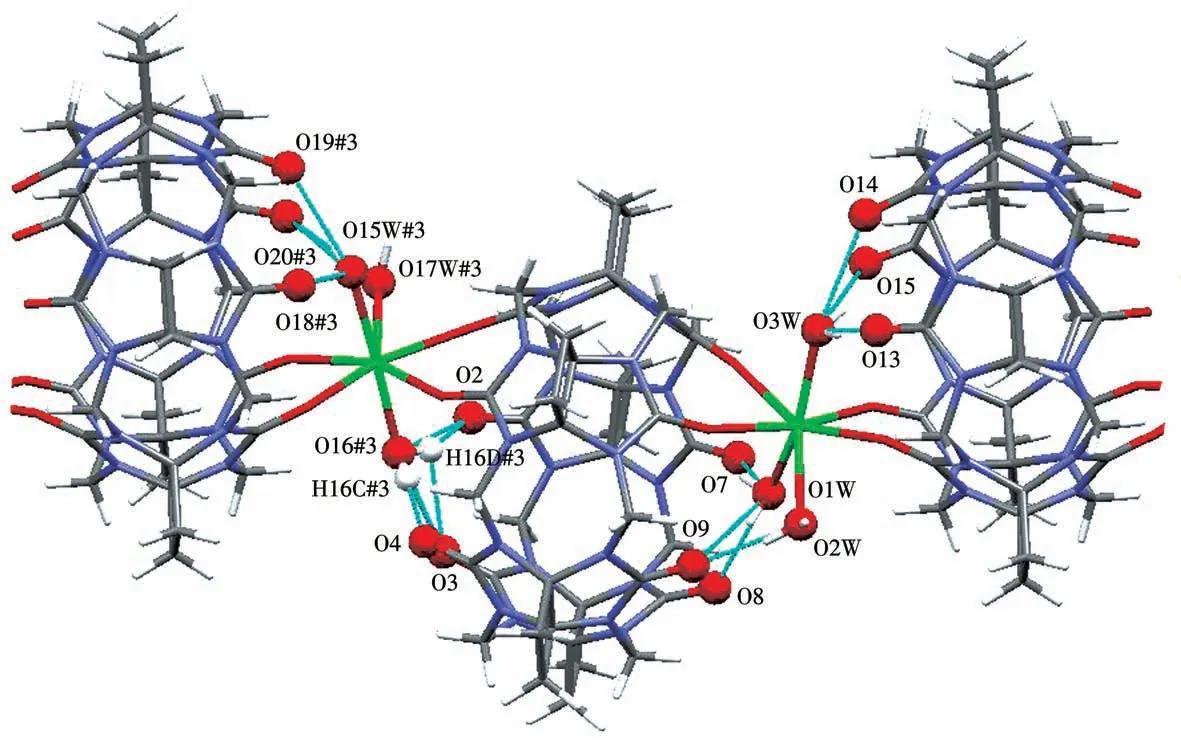
图9 化合物3的1D分子链中的非共价键相互作用Fig.9 Non-covalent bond interactions in the 1D molecular chains of compound 3
图10a为化合物3的3D堆积图,ClO4-、[ZnCl4]2-、Cl-和水分子填充在CyB5Q[5]-Ca的1D分子链间,形成不规则的孔洞结构。图10b为化合物3的1D分子链间的非共价键相互作用,2个链间填充了[ZnCl4]2-和ClO4
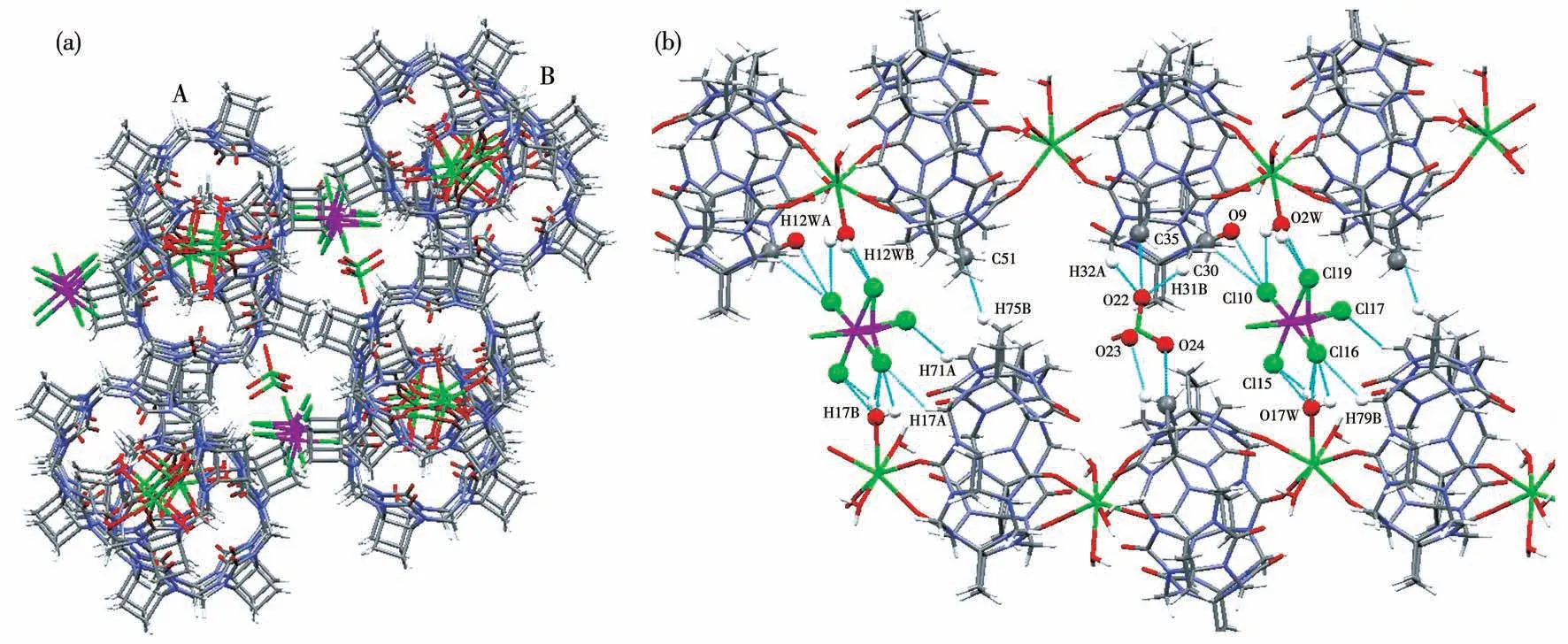
图10 (a)沿a轴观察的化合物3的3D堆积;(b)化合物3中A和B链间的非共价键作用Fig.10(a)Three-dimensional stacking of compound 3 viewed along the a-axis;(b)Non-covalent bonding interactions between the A and B chains in compound 3
-,并且2个阴离子分别以氧原子和氯原子通过偶极-偶极和氢键作用连接2个1D链,这些偶极-偶极作用包括O22…H31B(0.232 5 nm)、O22…H32A(0.244 2 nm)、O22…C35(0.306 1 nm)、O24…C11(0.315 4 nm)、O23…H11B(0.261 4 nm)、Cl15…O17W(0.273 1 nm)、Cl16…O17W(0.289 3 nm)、Cl17…H71A(0.288 4 nm)、Cl16…H79B(0.278 1 nm)、Cl10…O9(0.304 4 nm)、Cl10…H2WA(0.283 8 nm)、Cl15…O17W(0.273 1 nm)。氢键的类型和键长、键角见表2。
2.4 化合物4的晶体结构
图11为化合物4的晶体结构图。Ca1仅与CyB5Q[5]分子的一端2个羰基氧原子O3、O3#7配位,配位键长为0.237 6 nm;Ca1同时还与4个水分子O1W、O4W、O3W#7和O4W#7配位,配位键长分别为0.235 2、0.228 9、0.239 7和0.239 7 nm。O1W还与CyB5Q[5]端口羰基氧原子O1、O2和O2#7发生偶极-偶极作用,它们的作用距离分别为0.287 6、0.279 0和0.279 0 nm。钙离子与不同氧原子形成六配位结构,即Ca1和CyB5Q[5]形成一端封闭的“分子碗”(图11b)。化合物4的一维分子链(A、B链)间利用ClO4
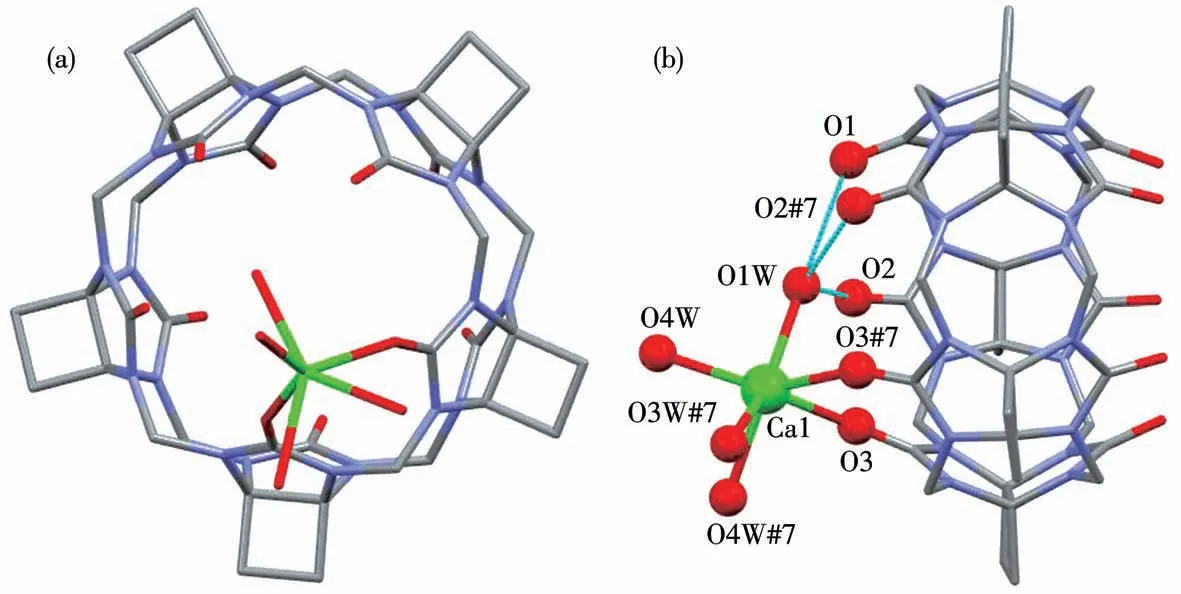
图11 化合物4的晶体结构图:(a)俯视;(b)侧视Fig.11 Crystal structure diagram of compound 4:(a)top view;(b)side view
-的2个 氧 原 子(O12、O12#1),分 别 与2个CyB5Q[5]分子的亚甲基氢(H13A、H14A和H13A#1、H14A#1)发生离子-偶极作用(图12c):O12…H13A和O12#1…H13A#1的 距 离 为0.251 8 nm,O12…H14A和O12#1…H14A#1的距离为0.240 9 nm;同时1D分子链间的相邻2个ClO4-间,利用O12、O12#1与O9形成2个偶极-偶极作用,它们间的作用距离均为0.291 6 nm;此外,1D链间的2个CyB5Q[5]分子还分别与临近的ClO4-形成2对氢键,这些氢键的键长、键角和细节分别见表1和图12c。化合物4依靠这些非共价键相互作用形成3D堆积,[ClO4]-和游离水分子堆积在CyB5Q[5]配合物的空隙中(图12a)。
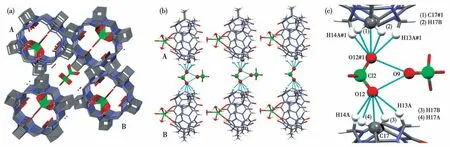
图12 化合物4的结构:(a)沿c轴观察的3D堆积图;(b)A、B链间的非共价键相互作用;(c)非共价键相互作用细节Fig.12 Structure of compound 4:(a)3D stacking viewed along the c-axis;(b)non-covalent bonding interactions between A and B chains;(c)details of the non-covalent bonding interactions
2.5 化合物1~4的结构表征
2.5.1 TGA和元素分析
图13a为化合物1的TG曲线。在109.2℃时,化合物失重11.10%,折合为8.1个游离水;250℃时,失重18.69%,折合为5.55个结合水,根据TG计算结果,可认为化合物1结构单元中共计有14个水分子。该化合物在440℃前都能稳定存在。再根据文献[21],CyB5Q[5]的分子式为C40H40N20O10,故化合物1的分子式为C40H68N20O24Cl2Ca(M=1 320.98),以此分子式所得的元素分析计算值(%):C 36.34,H 5.15,N 21.20;实测值(%):C 35.00,H 4.66,N 20.95。
图13b为化合物2的TG曲线。在109.2℃时,化合物失重8.10%,折合为6.30个游离水;250℃时,失重13.85%,折合为4.46个结合水,化合物2中共计有11个水分子。该化合物在440℃前都能稳定存在。同前述,其分子式确定为C40H62N20O21Cl4CaZn(M=1 403.26)。以此分子式所得的元素分析计算值(%):C 34.21,H 4.42,N 19.95;实测值(%):C 35.20,H 4.16,N 21.34。
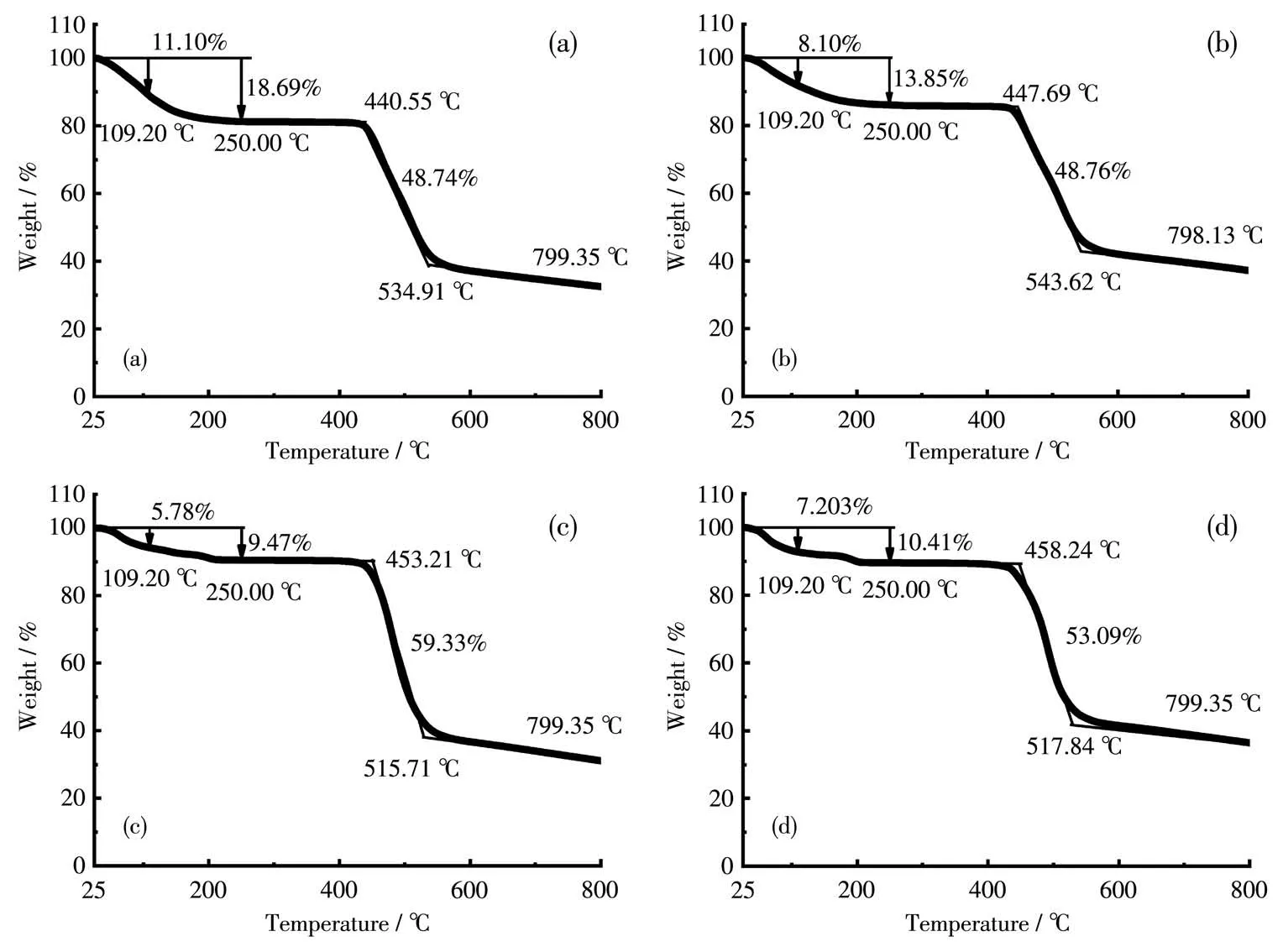
图13 化合物1~4的TG曲线(分别对应a~d)Fig.13 TG curves of compounds 1-4(a-d,respectively)
图13c为化合物3的TG曲线。在109.2℃时,化合物失重5.78%,折合为5.3个游离水;250℃时,失重9.47%,折合为3.35个结合水,故化合物3结构单元中共计有9个水分子。该化合物在440℃前都能稳定存在。其分子式确定为C40H61N20O23Cl9Ca2Zn(M=1 653.58)。以此分子式所得的元素分析计算值(%):C 29.06,H 3.57,N 16.95;实测值(%):C 29.19,H 3.89,N 17.32。
图13d为化合物4的TG曲线。在109.2℃时,化合物失重7.203%,折合为5.68个游离水;250℃时,失重10.41%,折合为2.5个结合水,故化合物4结构单元中共计有8个水分子。该化合物在440℃前都能稳定存在,其分子式确定为C40H58N20O26Cl4Ca(M=1 415.87)。以此分子式所得的元素分析计算值(%):C 33.90,H 4.10,N 19.78;实测值(%):C 33.25,H 3.94,N 21.15。
2.5.2 PXRD分析
对化合物1~4进行了PXRD分析,结果见图14。从配合物1~4的XRD实测结果与相应单晶数据模拟图比较能看到,配合物1~4的主要特征衍射峰位与由单晶数据理论拟合的峰位基本吻合,表明制备的化合物为纯相。强度与理论模拟图不太一致,这可能是由于晶体取向不同。
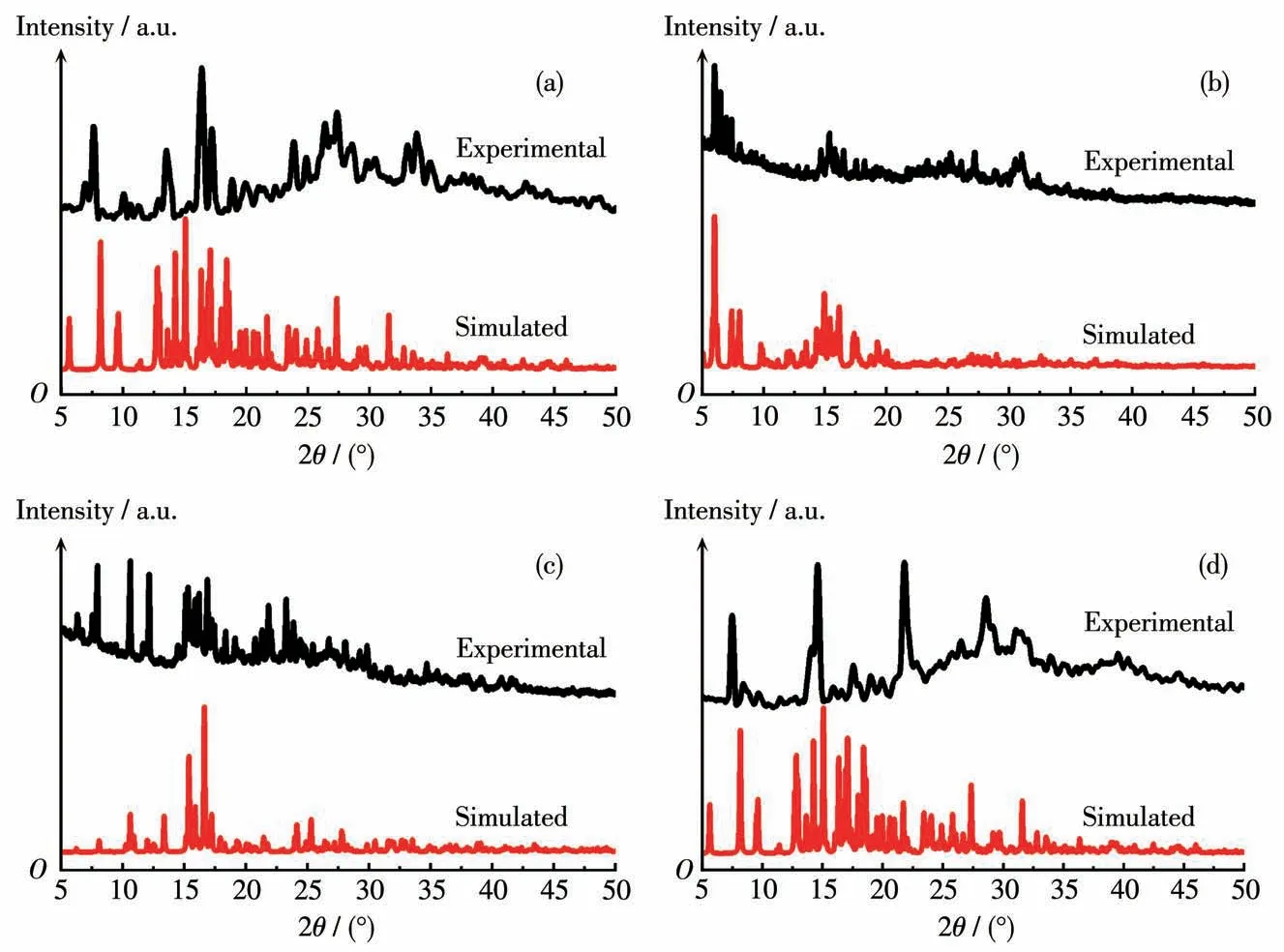
图14 化合物1~4的PXRD图(分别对应a~d)Fig.14 PXRD patterns of compounds 1-4(a-d,respectively)
3 结论
本文报道的4个化合物,都是由CyB5Q[5]与Ca生成的瓜环-Ca配位化合物。化合物1~3都是以钙离子与CyB5Q[5]的2个端口氧原子配位;同时,2个CyB5Q[5]分子间存在不同数目的与钙配位的水分子,其氧原子与CyB5Q[5]分子中未配位的端口氧原子通过非共价键作用将CyB5Q[5]两端封口,形成配位数分别为6、8和7的“分子胶囊”,即3种配合物都通过CyB5Q[5]-Ca-CyB5Q[5]配位键和其他非共价键相互作用连接为一维分子链(图3a、7a和10)。而化合物4中,钙离子除了与4个水分子的氧原子配位外,仅与CyB5Q[5]一端2个氧原子配位,形成6配位的“分子碗”,CyB5Q[5]分子中另一端未配位的氧原子和另一个CyB5Q[5]的配位水分子通过非共价键作用封口。化合物1的一维分子链间,主要以游离水分子、氯离子之间的非共价键相互作用堆积为超分子聚集体,而化合物2~4除了水分子、氯离子外,还分别以[ZnCl4]2-、ClO4-+[ZnCl4]2-和ClO4-与CyB5Q[5]发生非共价键相互作用,形成超分子聚集体(图4a、8a、11a和13a)。
由这些瓜环基超分子聚集体的晶体结构,可以发现,相同的金属阳离子(钙)和配体(CyB5Q[5])在不同的阴离子介质中,可能会形成结构不同的超分子金属有机骨架化合物,该现象是否可以作为合成瓜环基SOFs的普遍规律,将有待进一步的研究。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
发明与创新·中学生(2023年2期)2023-01-09 03:50:05
椰城(2021年12期)2021-12-10 06:08:52
中学生数理化(高中版.高二数学)(2020年6期)2020-07-24 08:18:22
数理化解题研究(2018年19期)2018-08-15 02:13:30
物理学报(2017年21期)2017-11-10 08:25:38
数理化解题研究(2016年28期)2017-01-04 07:01:21
腐蚀与防护(2016年7期)2016-09-14 09:30:56
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
无机化学学报(2014年1期)2014-02-28 17:30:06
