
NF-κB通路对口腔鳞癌细胞迁移和侵袭影响的初步研究
2022-09-28 13:46王爽邱林刘涵孙晓倩代晓华张军刘浩
实用口腔医学杂志 2022年5期
王爽 邱林 刘涵 孙晓倩 代晓华 张军 刘浩
口腔癌是头颈部最常见的恶性肿瘤,其中口腔鳞状细胞癌(oral squamous cell carcinoma,OSCC)占比90%以上[1-2],5 年生存率约50%左右,与其侵袭性强及淋巴结转移率高相关[3]。由于目前仍缺乏减少OSCC转移的有效方法,故探究其相关转移机制具有重要临床意义。
据报道,慢性炎症与OSCC发生和进展有关[4],而NF-κB通路作为炎症相关癌症调控中的经典通路,在包括OSCC在内的多数癌症中被异常激活[5]。该通路通过调控下游细胞因子和趋化因子表达来促使肿瘤细胞发生上皮间充质转化(epithelial-mesenchymal transition,EMT),随之产生许多促炎因子,因此建立一个调节环,有利于维持EMT表型[5]。尽管NF-κB通路已经发现数十年,但在OSCC中的作用尚不清楚。
1 材料与方法
1.1 细胞培养
本实验使用人舌鳞癌细胞系SCC-15(天津市口腔功能重建重点实验室存储),用含10%胎牛血清(Gibco,美国)和1%双抗溶液(北京Solarbio)的高糖DMEM,在37 ℃和5%CO2湿润培养箱中培养。
1.2 Transwell侵袭实验
预冷无血清DMEM按1∶3稀释Matrigel,取100 μL Matrigel胶均匀铺在Transwell小室内,37 ℃孵育4 h至凝结成胶冻状;设置20 μmol/L BAY11-7082预处理细胞1 h组、不预处理细胞组及PBS对照组,前两组皆用1 μg/mL LPS刺激饥饿12 h以上的细胞24 h后收集细胞。用无血清DMEM将收集的细胞浓度调整为5×105/mL,上室加200 μL细胞悬液,下室加600 μL含20%血清的DMEM,培养24 h;弃小室中培养液,4%多聚甲醛固定15 min,PBS漂洗2 次,DAPI室温避光染色5 min,倒置荧光显微镜下观察,每个小室随机拍摄5 个视野并计数。
1.3 裂痕愈合实验
细胞分组及处理同上,收集细胞后按7×104个细胞/孔接种到伤口愈合检测皿(ibidi, 德国)中,待细胞长满后拔掉皿中自带插件形成细胞铺展面裂痕,换成无血清培养基,初始伤口宽度为500 μm(0 h),细胞培养24 h,弃培养基,PBS漂洗后用倒置荧光显微镜采集裂痕宽度图像,Image J软件测量伤口闭合面积。
1.4 蛋白质免疫印迹(Western blot)
饥饿细胞12 h以上,用不同浓度的LPS处理细胞指定时间后收集细胞样品,等体积PBS作为对照。在4 ℃下提取细胞蛋白,BCA蛋白检测试剂盒测定蛋白浓度。用SDS-PAGE预制胶分离蛋白质,电转至PVDF膜,5%脱脂奶室温封闭1 h;用IκBα、p65、p-p65、ZEB2(1∶500,Santa Cruz,California,USA)、Tubulin、E-cadherin、Vimentin(1∶1 000,上海,Beyotime)4 ℃摇床孵育过夜;1×TBS漂10 min共3 次,HRP标记的山羊抗兔/小鼠IgG室温摇床(1∶1 000,Beyotime)孵育1 h,1×TBS漂10min共3次。化学发光试剂显色后用Image Lab自动凝胶分析软件对条带进行定量。
1.5 实时荧光定量PCR(real-time PCR)
饥饿细胞12 h以上,用1 μg/mL LPS处理不同时间段(0、0.5、1、2、3、4、5、6、8、16、24 h)后收集细胞,等体积PBS作为对照。提取细胞总RNA反转录后经Light Cycler/Light Cycler 480系统(罗氏,瑞士)进行RT-PCR扩增。GAPDH为内参,2-ΔΔCt法计算mRNA表达相对倍数变化。引物(中国生工生物技术)见表1。
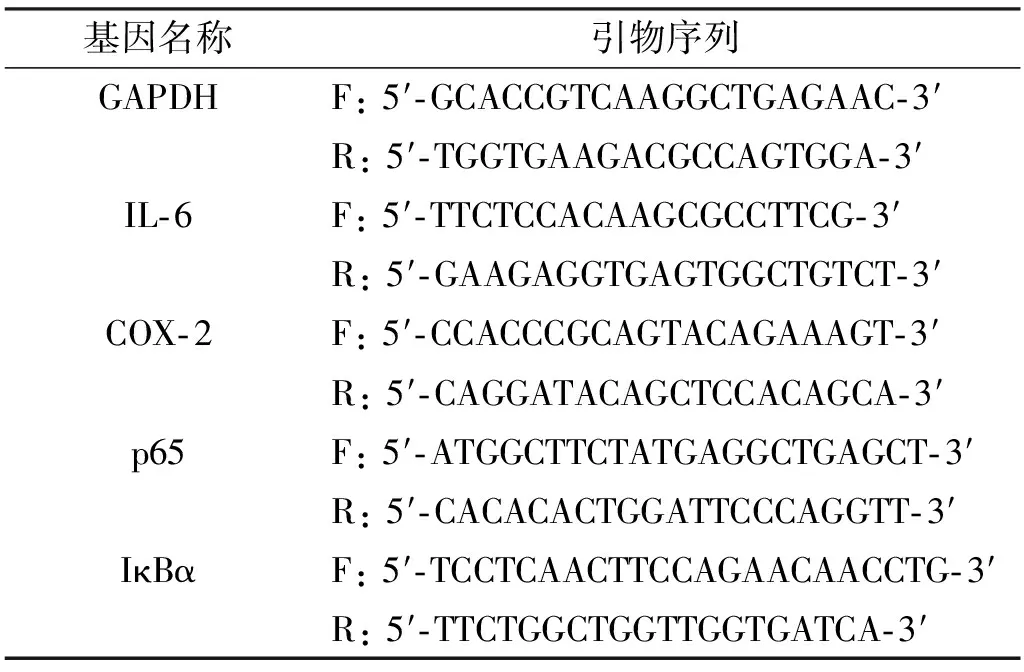
表1 引物序列
1.6 免疫荧光染色
饥饿细胞12 h以上,用1 μg/mL LPS刺激8 h后收集细胞,等体积PBS作为对照。按8×104个细胞/孔接种到共聚焦小皿中过夜后4%多聚甲醛固定15 min,PBS漂洗5 min共3 次;室温封闭15 min;4 ℃中与p65抗体孵育过夜后PBS漂洗15 min,室温下Cy3(红色)偶联荧光二抗(1∶200,Beyotime)避光染色1 h,PBS漂洗;DAPI避光染色5 min,PBS去浮色,倒置荧光显微镜对染色结果成像。
1.7 统计学分析

2 结 果
2.1 LPS激活SCC-15细胞NF-κB通路
SCC-15细胞经1 μg/mL LPS处理0、0.5、1、2、4、8、16和24 h后,IκBα 蛋白表达量在4 h到8 h显著降低(图1A);mRNA表达量在4~8 h显著升高(P<0.05),5 h时达到峰值,之后逐渐下降(图1B)。结果提示,IκBα作为NF-κB通路的负反馈调节因子,在LPS刺激SCC-15细胞4 h后蛋白表达水平降低,从而激活NF-κB通路;免疫荧光结果显示,对照组中p65核中染色弱(红色荧光),而处理组p65核中染色强(图1C),LPS诱导 p65从细胞质向核转运。上述结果表明LPS成功诱导NF-κB通路活化。
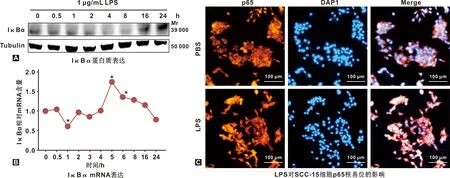
图1 LPS激活SCC-15细胞的NF-κB通路
2.2 NF-κB通路调控SCC-15细胞迁移和侵袭
伤口愈合实验显示,相比对照组,SCC-15细胞迁移能力提高了1.785 倍(P<0.05,图2A);Transwell侵袭实验中,相比对照组,SCC-15细胞侵袭能力提高56.4%±7.2%(P<0.05,图2B)。但NF-κB通路抑制剂BAY11-7082处理细胞后,相比LPS处理组,BAY11-7082组显著降低SCC-15细胞的相对迁移面积(P<0.05,图2C)以及穿膜细胞数量,平均每个视野中穿膜细胞数不足LPS组的1/4(P<0.05,图2D)。结果表明,NF-κB通路活化促进SCC-15细胞迁移和侵袭。
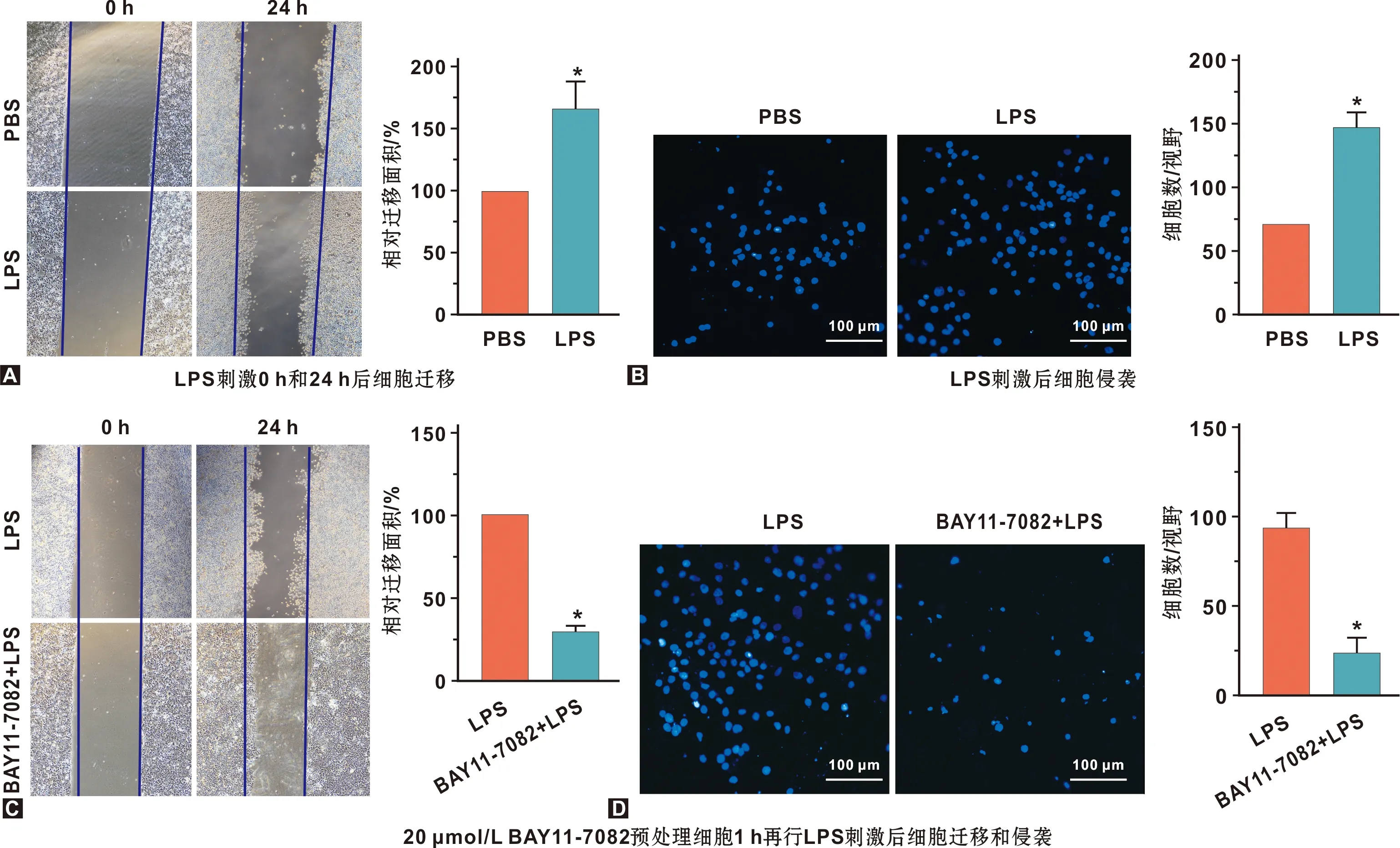
图2 NF-κB通路调控SCC-15细胞迁移和侵袭
2.3 LPS上调SCC-15细胞中p65磷酸化蛋白及促炎因子
p65 mRNA表达水平在经LPS刺激0、0.5、1、2、3、4、5、6、8、16和24 h后均无明显变化(P>0.05, 图3A);而p65磷酸化蛋白表达水平分别呈LPS刺激时间和浓度依赖性,24 h达峰值,较对照组上调3.39 倍,0.1 μg/mL LPS 处理下即可显著上调其磷酸化蛋白,1 μg/mL和10 μg/mL LPS处理时上调更为显著(图3B~3C)。这表明,NF-κB通路活性可能与SCC-15细胞中p65磷酸化蛋白水平相关。
通过RT-PCR检测相同刺激条件下IL-6和COX-2 mRNA表达,发现相比对照组,处理组显著性上调IL-6和COX-2 mRNA表达水平(P<0.05)。其中,IL-6在LPS处理0.5 h后达到峰值(3.71 倍),随后上调量逐渐减少,5 h后恢复至对照组水平(图3D);COX-2在LPS处理0.5 h后显著上调,至5 h达到峰值(10.2 倍),之后缓慢降低,24 h时恢复至对照组水平(图3E)。上述结果提示LPS促进SCC-15细胞中促炎因子表达。
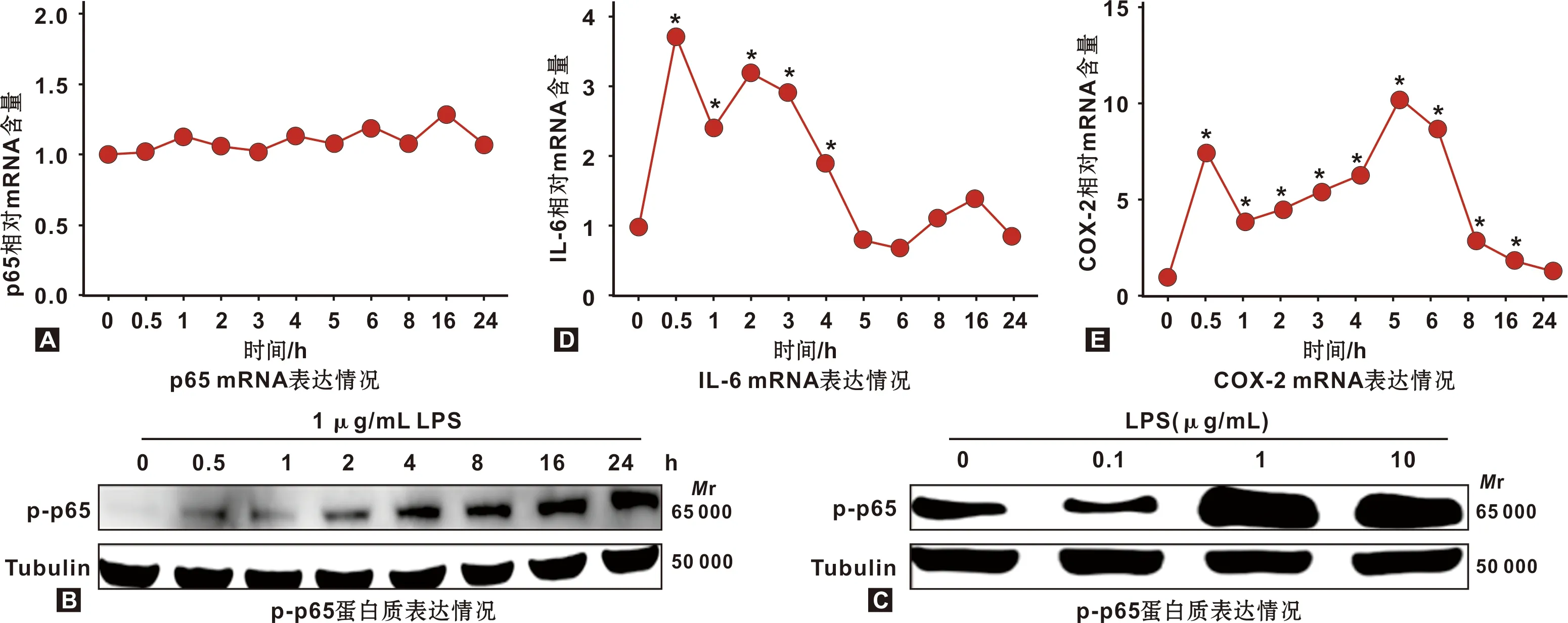
图3 LPS上调SCC-15细胞中p65磷酸化蛋白及促炎因子
2.4 LPS促进SCC-15细胞EMT
浓度为1 μg/mL以上的LPS处理SCC-15细胞24 h后,相比对照组,处理组上皮标记物E-cadherin表达显著降低,而间充质标记物Vimentin和ZEB2表达水平显著升高(P<0.05,图4),表明LPS促进SCC-15细胞发生EMT。
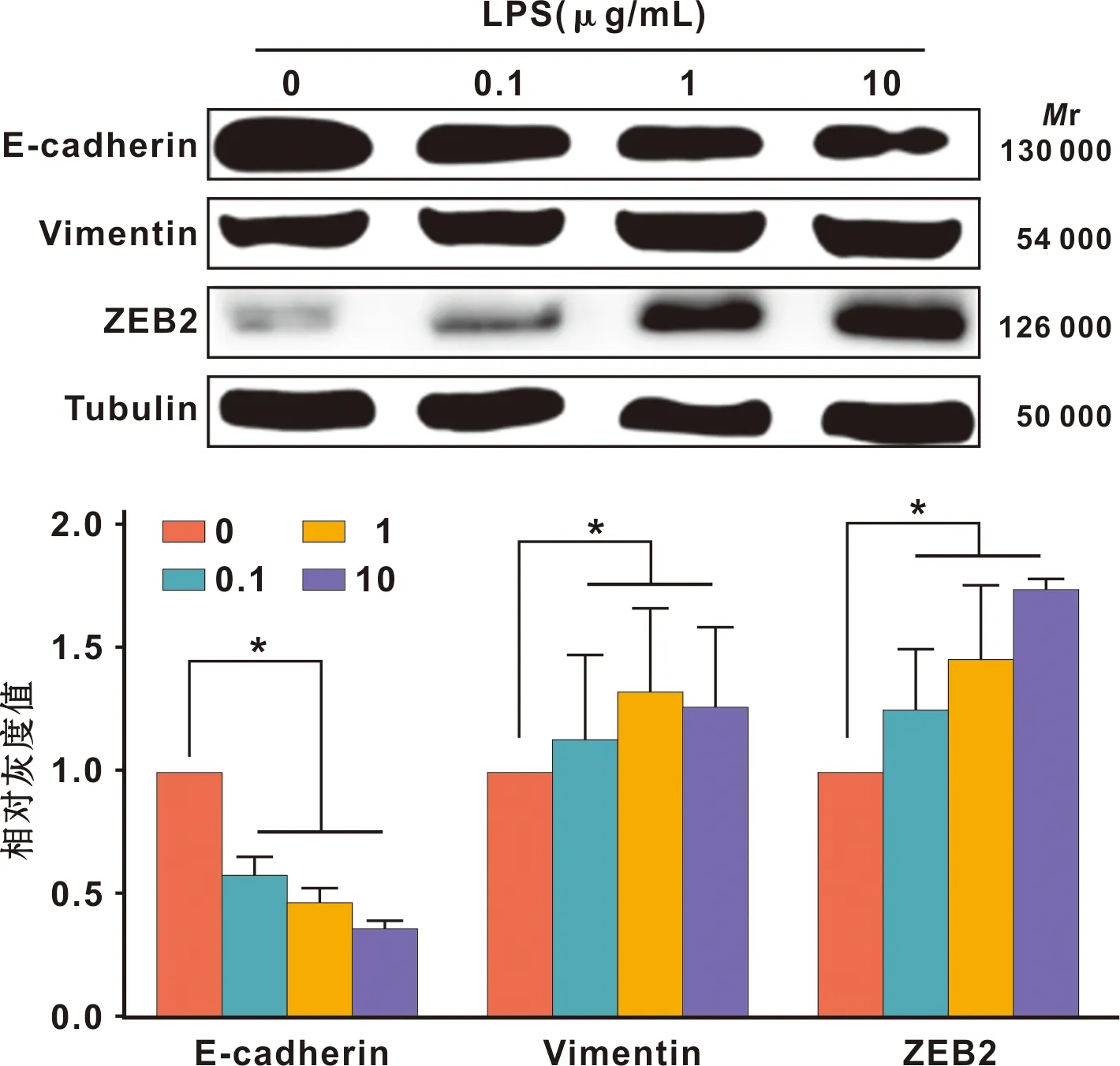
图4 LPS促进SCC-15细胞EMT
3 讨 论
目前已知NF-κB家族由5 个成员组成:RELA(p65)、RELB、c-REL、p50/p105(NF-κB1)和p52/p100(NF-κB2),它们可以形成二聚物复合物。这些复合物被其抑制蛋白(IκBs)限制在胞质中,可被各种刺激(如细胞因子、细菌感染等)激活易位至胞核,进而激活下游促炎因子(如IL-6和COX-2等)[6]。本实验结果显示,经LPS诱导后,p65在胞核中明显表达,且SCC-15细胞中IκBα mRNA在4~6 h再合成,蛋白在4~8 h显著降解,这表明IκBα mRNA可翻译成活性的IκBα蛋白,蛋白降解和再合成的量相当,而在8 h后IκBα蛋白维持在对照组水平;同时p65(Ser311)磷酸化蛋白水平呈持续性上调,这说明NF-κB通路持续响应LPS刺激而处于转录激活状态。
NF-κB是参与炎症相关癌症调节的中心信号通路,该通路的异常活化能够加速癌症发展[7]。IL-6具有肿瘤生长促进作用,在OSCC中有明显的高表达且与p65的表达呈正相关[8]。相比于正常口腔黏膜,OSCC中COX-2高表达并与肿瘤局部复发、淋巴结转移有关;同时COX-2可上调细胞间粘附分子-1(ICAM-1)和血管内皮生长因子C(VEGFC),进而促进OSCC转移[9-10]。在本研究中,LPS激活组中SCC-15细胞迁移和侵袭性较对照组明显增强,而用BAY11-7082抑制NF-κB通路后,细胞迁移和侵袭性明显减弱,这表明NF-κB通路可能调节OSCC细胞迁移和侵袭。
恶性肿瘤转移是一个多步骤过程[11],其中通过EMT过程能使细胞间粘附丧失,同时能促进迁移和侵袭所需的形态和行为转化[12]。炎症中的可溶性因子可以促进癌细胞获得EMT相关的运动性、侵袭性和降解细胞外基质等特征[13]。在本研究中,经LPS刺激,发现维持正常上皮细胞间粘附的E-cadherin表达显著下调,同时Vimentin和ZEB2表达显著上调,由于Vimentin的高表达与OSCC转移潜力增加相关[14],且NF-κB可通过刺激转录因子ZEB2转录来抑制E-cadherin表达[15],因此使得SCC-15细胞具有更强的迁移、侵袭潜能,表明激活的NF-κB通路可能通过调节EMT过程促进OSCC迁移和侵袭。
总之,本文通过体外研究初步证实炎症通路NF-κB可以调节EMT过程且促进OSCC迁移和侵袭,为阐述NF-κB通路在OSCC中的作用提供部分实验证据。
猜你喜欢
生殖医学杂志(2022年10期)2022-10-19
西北民族大学学报(自然科学版)(2022年2期)2022-07-06
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
中风与神经疾病杂志(2021年9期)2021-11-08
纺织科技进展(2021年4期)2021-07-22
兰州理工大学学报(2021年3期)2021-07-05
中小学德育(2020年11期)2020-03-18
分析化学(2017年12期)2017-12-25
中国市场(2017年5期)2017-03-15
