
铂单原子催化剂同步辐射X射线吸收谱的研究进展
2022-09-19 06:28任诗杰谯思聪刘崇静张文华
高等学校化学学报 2022年9期
任诗杰,谯思聪,刘崇静,张文华,宋 礼
(1.中国科学技术大学国家同步辐射实验室,2.化学与材料学院,合肥 230029)
为了应对日益增长的能源需求和由化石燃料消耗引起的环境问题所带来的挑战,研发基于电化学、光化学和热化学的新型清洁能源储存和转化体系势在必行[1~3].近年来,一些涉及氢气析出、氢气氧化、氧气还原和费托合成[4]等反应的能源器件,由于可以产生高能量密度的化学燃料或实现高效电储存而被广泛关注.然而,这些反应过程往往面临着能量转化效率低、产物选择性差和动力学过程缓慢等问题,严重阻碍了其商业化大规模应用[5].催化剂的使用可以促进电子质子耦合,从而降低整体反应的动力学势垒,同时通过调节中间体吸附行为来抑制潜在的副反应,使得反应转化效率和选择性显著提高[6~8].因此,制备高活性和稳定性的催化剂对于这些能源储存和转化设备的优化设计至关重要.
在众多异质催化剂中,铂(Platinum,Pt)基催化剂具有本征催化活性高、中间体吸附强度适宜和能带结构可调等优势,近年来逐渐成为了研究热点之一[9,10].原子化[11]、界面作用[12]、应力工程[13]和配体修饰[14]等策略可通过调控Pt位点的电子结构和配位构型,来进一步提高其催化活性.其中,原子化策略将Pt以单个原子形式分散在适当载体上来合成单原子催化剂,实现每个Pt原子都作为活性位点,使得催化性能显著提升,并有效节省了贵金属资源[9,10,15].同时,得到的Pt单原子催化剂具有结构高度均一性,可以作为理想的模型催化剂应用于反应机理的研究.
另一方面,纳米碳[16]、金属氧化物[17,18]、金属单质[19]和多孔有机框架[20]等单原子催化剂载体,不仅可以通过配位键合Pt原子来抑制金属偏析,还可以通过适当的耦合效应改变单分散Pt位点的电子特性,对于催化活性具有重要的调控作用[17].因此,如何更深入地理解载体与孤立Pt位点之间的相互作用,建立催化剂结构性能关联机制,是进一步促进单原子催化剂发展的关键所在[21~23].然而,常规的表征技术在研究单原子催化剂的局域结构和微反应环境时具有局限性,这使得建立催化剂的真实反应模型极具挑战.
近年来,逐渐发展和完善的多种原位、非原位同步辐射技术[24]为精确表征单原子催化剂局域结构、揭示金属载体耦合作用机制和追踪催化剂在反应过程中的动态演化过程提供了更多可能[25,26].如,X 射线吸收谱(X-Ray absorption spectroscopy,XAS)分为X 射线吸收近边结构谱(XANES)和扩展X 射线吸收精细结构谱(EXAFS),其能够提供配位结构和电子跃迁等信息;同步辐射光电子发射谱(Synchrotron radiation photoemission spectroscopy,SR-PES)可以表征金属原子的氧化态;同步辐射傅里叶变换红外光谱(Synchrotron-radiation Fourier-transform infrared spectroscopy,SR-FTIR)被用来探测单原子催化过程中的关键吸附物种等.
基于此,本文综合评述了结合同步辐射技术研究不同载体负载Pt单原子催化剂的最新进展,讨论了非原位和原位同步辐射X射线吸收谱在金属-载体相互作用解析、真实催化位点指认和构效关系理解中的应用,并对其面临的挑战和发展前景进行了展望.
1 XAS的原理及装置
当X射线穿过材料时会因为吸收而发生强度衰减,根据比尔定律,这种衰减可以通过吸收系数来表示[27]:
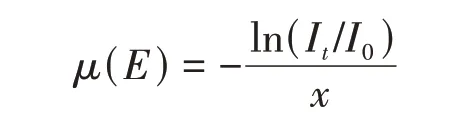
式中:I0和It分别为入射X射线和透射X射线的强度;x(mm)为样品厚度;μ(E)是依赖于光子能量的吸收系数.XAS就是基于X射线吸收系数的能量依赖性精细结构[28~30].吸收系数与入射X射线能量之间的关系受基本化学状态和局部环境的影响,可分为边前、边后和扩展边区域[31].当入射X 射线能量低于电子结合能时,电子不会被激发到高能非占据轨道或真空[32,33];此时X 射线与电子的弱相互作用导致μ(E)-E谱中出现平坦区域;一些符合跃迁定则的较低能跃迁也可在此区域表现为边前峰[图1(A)][28~30,34].当X射线能量足够高时,芯电子被激发至高能非占据轨道,μ(E)显著增大,边前和近边区对被检测元素的氧化态和电子水平敏感,因此一起称为XANES[28~30,34~37].XANES包含丰富的结构信息,可以从配位化学、分子轨道、能带结构和多重散射等方面进行定性描述[28~38].进一步增加X射线能量,被激发到连续态的出射和被散射电子的波函数与吸收原子的波函数相互作用,出现EXAFS[39,40];对EXAFS 进行最小二乘拟合[41,42],可以得到吸收原子的键长和配位数等局域配位信息[图1(B)][43].
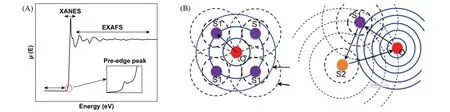
Fig.1 Typical XAS spectra(A)[30],the schematic illustration of the single(left) and multiple(right)scattering process of the excited photoelectrons(B)[43]
XAS信号的收集有透射、荧光和全电子产额3种基本模式[29,34,37].其中,透射模式测量入射和透射X射线强度之间的差异;荧光模式测量电子退激产生的X射线荧光;全电子产额则检测二次电子、俄歇电子等信号[图2(A)].另一方面,近年来逐步发展和完善的原位XAS测试装置,使得追踪目标元素在反应过程中电子结构和局域配位的动态演化过程成为了可能[44~46].图2(B)和(C)给出了一种典型的电化学原位池,其含有4个气孔,可用于研究涉及气体的反应;工作电极碳纸被胶带固定在前窗上,以防止电解质泄漏[30].该原位池可容纳30 mL液体.

Fig.2 Schematic diagram of three XAS detection modes(A),a photo of in situ XAS experiment setup(B)and diagram of the in situ cell(C)[30]
2 金属氧化物负载Pt单原子催化剂及其同步辐射XAS
金属氧化物是单原子催化剂中最常用的载体之一[47,48],其表面通常含有丰富的台阶位、边角位、氧空位和金属空位等缺陷位点,可以有效锚定高表面能的单分散Pt 物种[49,50].2011 年,Zhang 课题组[51]利用共沉淀法制备了Pt1/FeOx单原子催化剂;占据Fe 位置的Pt 原子促进了Fe2O3上氧空位的形成,从而大大提高了载体的还原性.另一方面,金属与载体之间的强相互作用(MSI)可以充分抑制Pt 单原子团聚以及调节Pt 位点的催化性能,这为氧化物基Pt 单原子催化剂的设计提供了一种新的思路[52~54].基于此,Wang 等[55]运用高温煅烧法合成了Pt1/Fe2O3单原子催化剂;在800 ℃下,PtO2从纳米颗粒(NPs)中蒸发,与还原性的Fe2O3作用形成Pt—O—Fe 共价键.球差校正透射扫描电子显微镜和原位红外光谱表明,相比不可还原的Al2O3负载Pt NPs,Pt1/Fe2O3表现出良好的单分散特性.同时,他们还用湿浸渍法(IWI)获得了负载量高达5%(质量分数)的Pt单原子催化剂.这些结果证明,设计适宜的共价金属-载体的相互作用(CMSI),有利于高负载量、耐高温的Pt 单原子催化剂的可控合成[52~55].
同步辐射XAS可以准确反馈单原子催化剂的电子结构和局域配位特征,从而为理解反应过程和单原子催化剂的优化设计提供指导[56,57].Narula 团队[58]通过湿化学法合成了Pt/θ-Al2O3单原子催化剂;Pt L3边XANES谱表明,0.18%(质量分数)Pt/θ-Al2O3的白线峰强度接近PtO2,说明样品中Pt以高氧化态存在[图3(A)].在5%H2/He气氛、150 ℃下反应60 min,0.18%(质量分数)Pt/θ-Al2O3的白线强度几乎无变化,暗示了氧化态Pt得到保持[图3(B)].EXAFS拟合指出,Pt—O的配位数和键长在还原处理前后几乎不变,与XANES分析结果一致[图3(C)和(D)].类似的,Lu等[59]利用原子层沉积(ALD)将原子分散的Pt 负载在Co3O4,ZrO2和CeO23 种氧化物载体上.如图4(A)所示,Pt1/Co3O4,Pt1/ZrO2和Pt1/CeO2中Pt均以氧化态存在.EXAFS显示3种Pt单原子催化剂中均出现了明显的Pt-O配位;同时Pt1/Co3O4中出现了Pt-Co信号,反映了Pt单原子与Co3O4载体之间的强相互作用[图4(B)~(D)].

Fig.3 Pt L3⁃edge XANES spectra of 0.18%(mass fraction) Pt/θ⁃Al2O3(A),XANES spectra before and after reduction treatment(B),EXAFS spectra(C)and EXAFS results before and after reduction(D)[58]
另一方面,利用原位XAS 谱检测局域结构动态演化,对于鉴别反应中间过程的真实活性位点结构至关重要.Newton等[60]结合质谱(MS),漫反射红外光谱(DRIFTS)和XAS研究了Pt/Al2O3催化CO低温氧化的反应过程,结果表明,Pt纳米颗粒表面吸附的CO转移至Pt单原子上形成碳酸根物种,从而实现了CO的储存与转化.XANES指出,Pt的L3边谱线强度只发生了很小程度的变化,证实了部分Pt表面发生可逆氧化[图5(A)和(B)].
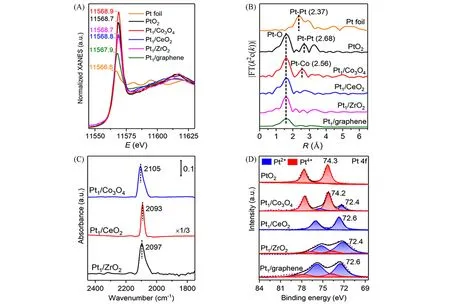
Fig.4 Pt L3⁃edge XANES spectra of Pt1/Co3O4,Pt1/CeO2,Pt1/ZrO2,Pt1/graphene,Pt foil and PtO2(A),the corresponding EXAFS spectra(B),DRIFTS spectra of Pt1/Co3O4,Pt1/CeO2 and Pt1/ZrO2(C),Pt4f XPS spectra of PtO2,Pt1/Co3O4,Pt1/CeO2,Pt1/ZrO2 and Pt1/graphene(D)[59]

Fig.5 Normalized Pt L3⁃edge XANES spectra(A) and temporal variation of Pt L3 white line intensity during a single cycle of redox operation(B)[60]
3 金属负载Pt单原子催化剂及其同步辐射XAS
近年来,由于同时保持了单原子和合金材料的优势,通过在传统金属催化剂表面引入单分散位点制备的单原子合金催化剂(Single-atom alloys,SAAs)逐渐成为了研究的前沿之一[61].孤立金属位点和金属载体之间的类合金相互作用引起催化剂电荷极化,从而有效调节了反应物种的吸附行为,优化了催化活性[62].多种策略已被报道用来可控制备Pt 单原子合金催化剂;如,Sun等[63]利用原子稀释的方法合成了Pt/Cu 单原子合金,Pt 优先分散在Pt/Cu 合金的表面层.Huang 等[64]利用局部电化学去合金法,在Pt 纳米线上形成Ni 单原子修饰层(SANi-PtNWs).高角度环形暗场扫描透射电子显微镜(HAADFSTEM)和电感耦合等离子体原子发射光谱(ICP-AES)证明,每平方纳米上有2.4个Ni原子和15个Pt原子,即表面Ni和Pt的原子比约为1∶6[图6(A)].电子能量损失谱(EELS)结果进一步证明了Ni的高分散度[图6(B)和(C)].同步辐射EXAFS指出没有Ni—Ni键存在,Ni以单原子形式分散在Pt纳米线表面[图6(D)和(E)].SANi-PtNWs表现出优异的析氢反应(HER)催化活性,其过电位、Tafel斜率和质量活性均优于商业Pt/C催化剂[图6(F)~(H)].Pan等[65]通过氨硼烷还原法合成了一种金属有机框架(MOF)稳定的PtPd 单原子合金光催化剂(Pd10@Pt1/UiO-66-NH2).HAADF-STEM 照片以及EXAFS 图中不存在Pt—Pt散射,证明了Pt以单原子形式分散在Pd纳米颗粒表面上[图7(A)和(B)].Pt与Pd的合金化导致Pt上电子聚集,促进了Pt位点的光催化析H2催化活性的提升.
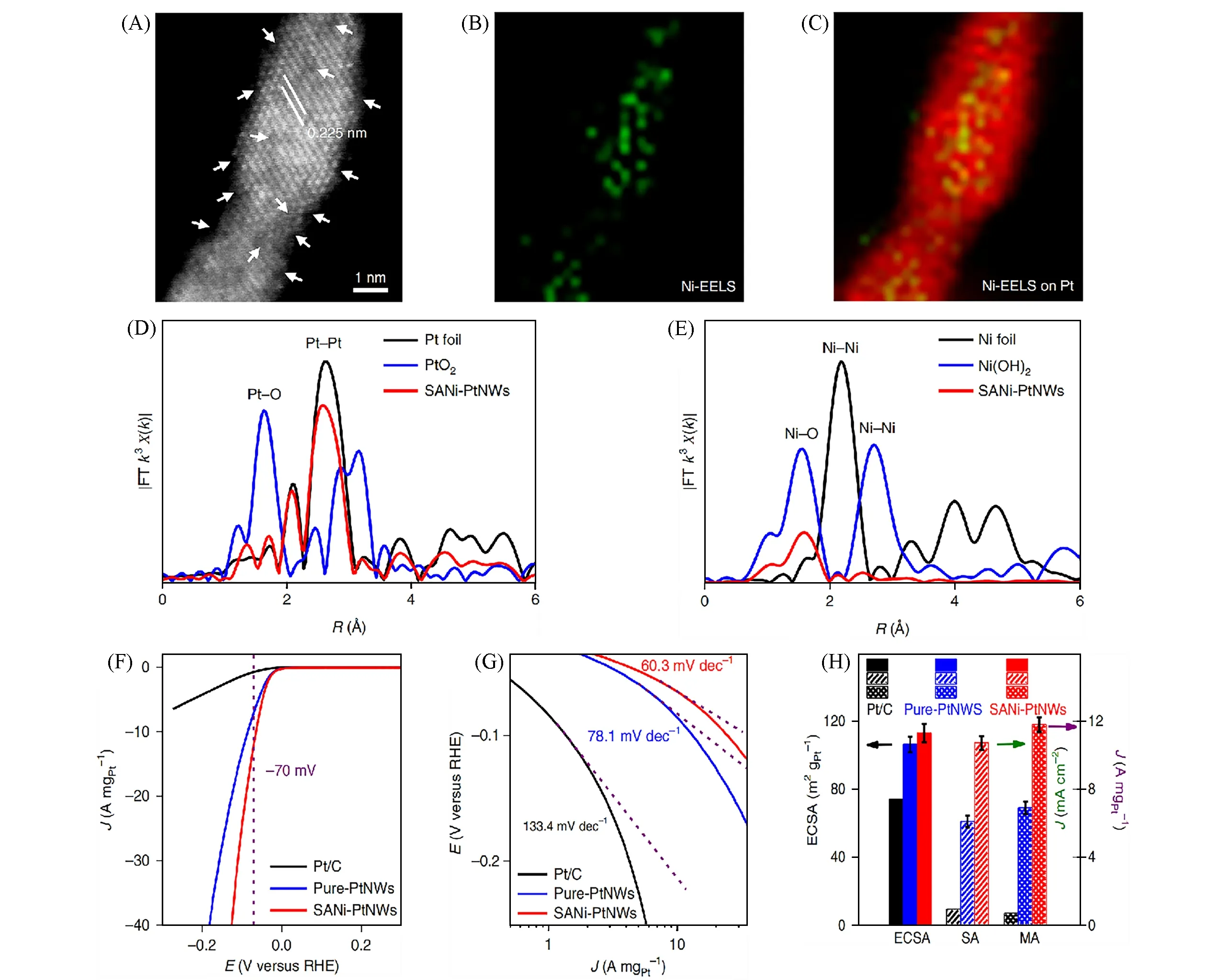
Fig.6 HAADF⁃STEM image of SANi⁃PtNWs(A),EELS mapping images(B,C),Pt L3⁃edge EXAFS spectra of Pt foil,PtO2,SANi⁃PtNWs(D),Ni K⁃edge EXAFS spectra of Ni foil,Ni(OH)2,SANi⁃PtNWs(E),HER LSV curves(F) and Pt mass normalized HER Tafel slope of Pt/C,pure⁃PtNWs,SANi⁃PtNWs(G),comparison of ECSA,specific activity(SA)and mass activity(MA)values at −70 mV(vs.RHE)of Pt/C,pure⁃PtNWs,SANi⁃PtNWs(H)[64]
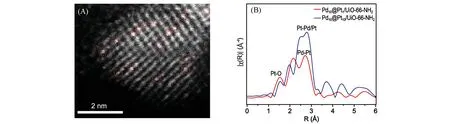
Fig.7 HAADF⁃STEM image of Pd10@Pt1/UiO⁃66⁃NH2(A)and the corresponding EXAFS spectra(B)[65]
另一方面,引入孤立金属位点所致的应变效应也可以通过调节电子结构来显著影响材料的催化性能.Wan 等[66]通过改变Pt 和Co 前驱体用量,合成了一系列具有不同壳层厚度和晶格应变程度的Au@(PtCo0.05)x核壳催化剂.Pt-Co壳体越厚,晶体中的机械弛豫导致Pt-Co壳层上的拉伸应变越小.在同步辐射XANES 谱中,Au@Pt1.5Co0.08的白线强度略高于Au@Pt1.5Co0.08和Au@Pt1.5,说明由于Au 核和Pt-Co壳层之间的强相互作用,Au@Pt1.5Co0.08的5d轨道填充较少,并且Co和Pt原子之间的d轨道杂化更强,从Pt原子到Co原子的电荷转移更多.因此,Au诱导的Pt-Co壳层拉伸应变促进了电子从Pt向Co的转移,从而降低了Pt的d带[图8(A)~(D)].
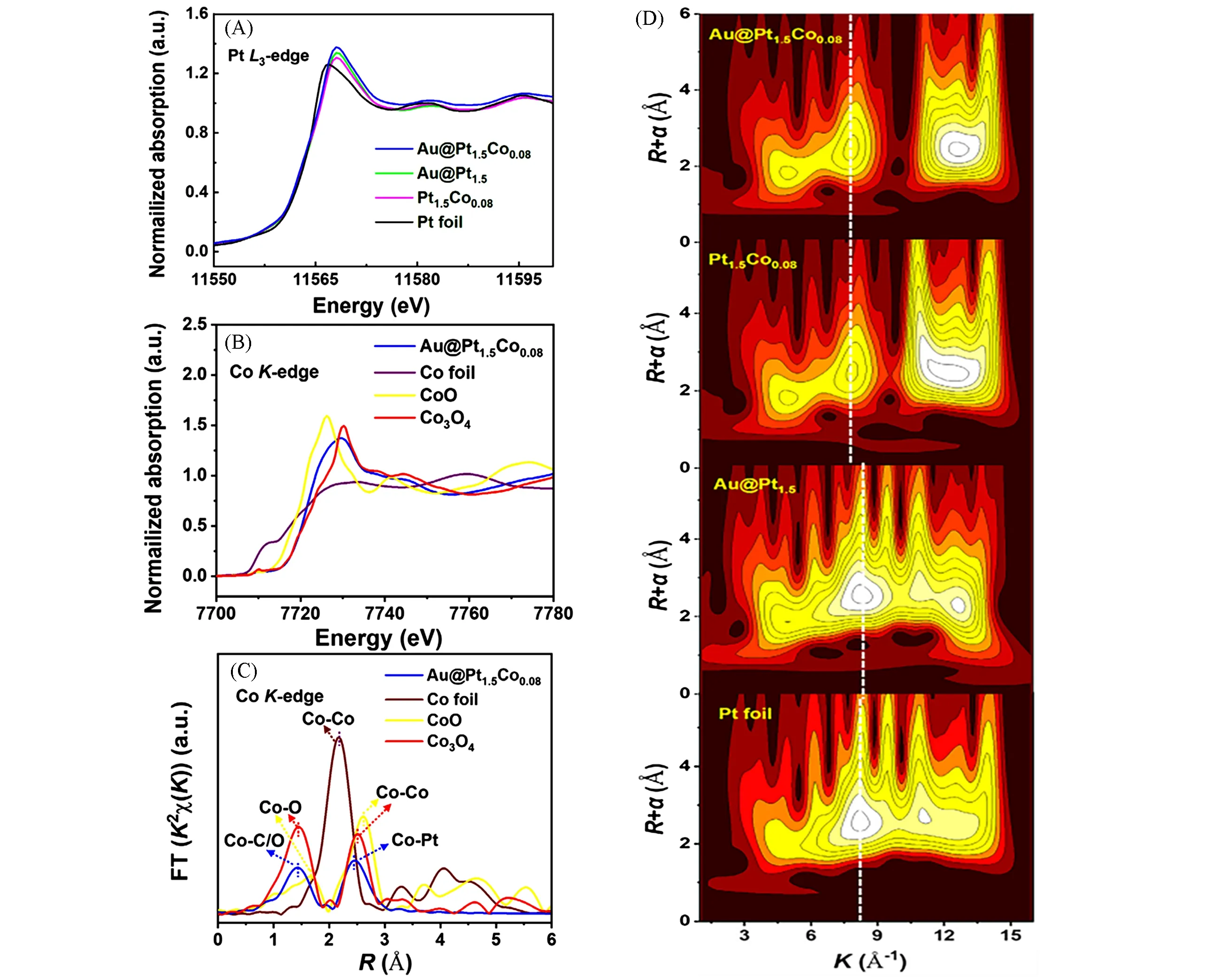
Fig.8 Pt L3⁃edge XANES spectra of Au@Pt1.5Co0.08,Au@Pt1.5,Pt1.5Co0.08,Pt foil(A),Co K⁃edge XANES spectra(B) and Co K⁃edge EXAFS spectra of Au@Pt1.5Co0.08,Co foil,CoO,Co3O4(C),wavelet transform(WT) EXAFS spectra of Au@Pt1.5Co0.08,Pt1.5Co0.08,Au@Pt1.5 and Pt foil(D)[66]
4 纳米碳负载Pt单原子催化剂及其同步辐射XAS
纳米碳材料由于具有高活性、优异的导电性和强结构可操作性而被广泛应用于催化领域.研究结果表明,在碳材料中引入的缺陷、掺杂异质原子和表面功能基团等可以作为单原子锚定位点,有效地抑制了金属偏析.如Ma等[67]利用缺陷捕获策略合成了具有Pt-C4配位结构的Pt单原子电催化剂.Song课题组[68]提出了不同维度的碳载体对催化性能也有影响,并设计及制备了以零维碳纳米洋葱为载体的Pt1/OLC单原子催化剂[图9(A)和(B)].
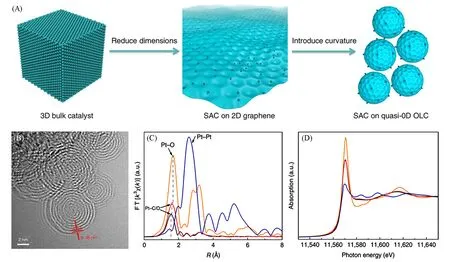
Fig.9 Schematic synthesis diagram of Pt1/OLC(A),TEM image of Pt1/OLC(B),Pt L3⁃edge FT⁃EXAFS spectra of Pt1/OLC(red),along with PtO2(yellow),Pt foil(blue) and Pt ligands/OLC(black)(C),the corresponding normalized XANES spectra(D)[68]
同步辐射EXAFS 谱图表明,Pt1/OLC 在1.67 Å(1 Å=0.1 nm)的位置出现了一个较强的峰,与PtO2中Pt—O键的位置类似;同时,对应Pt—Pt键的位置未出现较明显的峰,说明Pt以单原子形式负载在碳纳米洋葱之上[图9(C)].另外,Pt1/OLC 的峰强相对于未去配体的Pt ligands/OLC 有所增加,并且峰位向PtO2中Pt—O 的位置移动,说明在去配体过程中有新的Pt—O 键形成.进一步分析相应的Pt L3边XANES 谱,Pt1/OLC 峰的白线位置在Pt foil(Pt0)与PtO2(Pt4+)之间,充分说明了Pt 单原子处于氧化态[图9(D)].
相比复杂的碳缺陷构筑过程,通过杂原子掺杂来引入单原子锚定位点是一种更易于操作的方法[69~72].杂原子掺杂位点不仅可以与金属原子配位来实现高稳定性,而且可以充分调节金属的电子结构[73~75].由于与碳的相似性与相容性,具有吡啶N、吡咯N、石墨N、季N 和腈N 等多种构型的氮原子[76],是单原子催化剂中使用最广泛的配位位点.Sun 等[77]利用原子层沉积(ALD)技术制备了一种高稳定性、高活性的氮掺杂石墨烯负载Pt 单原子催化剂(Pt/N-graphene),HER 性能测试结果是商业Pt/C 催化剂的37倍[图10(A)~(C)].结合同步辐射XAS、HAADF-STEM 和密度泛函理论(DFT),发现氮原子的引入增强了Pt 原子和载体之间的相互作用,从而使Pt 锚定在N-graphene 载体表面,并且在催化过程中能够始终保持原子级分散;同时Pt 原子和载体N原子之间杂化产生了独特的5d电子结构[图10(D)~(G)].
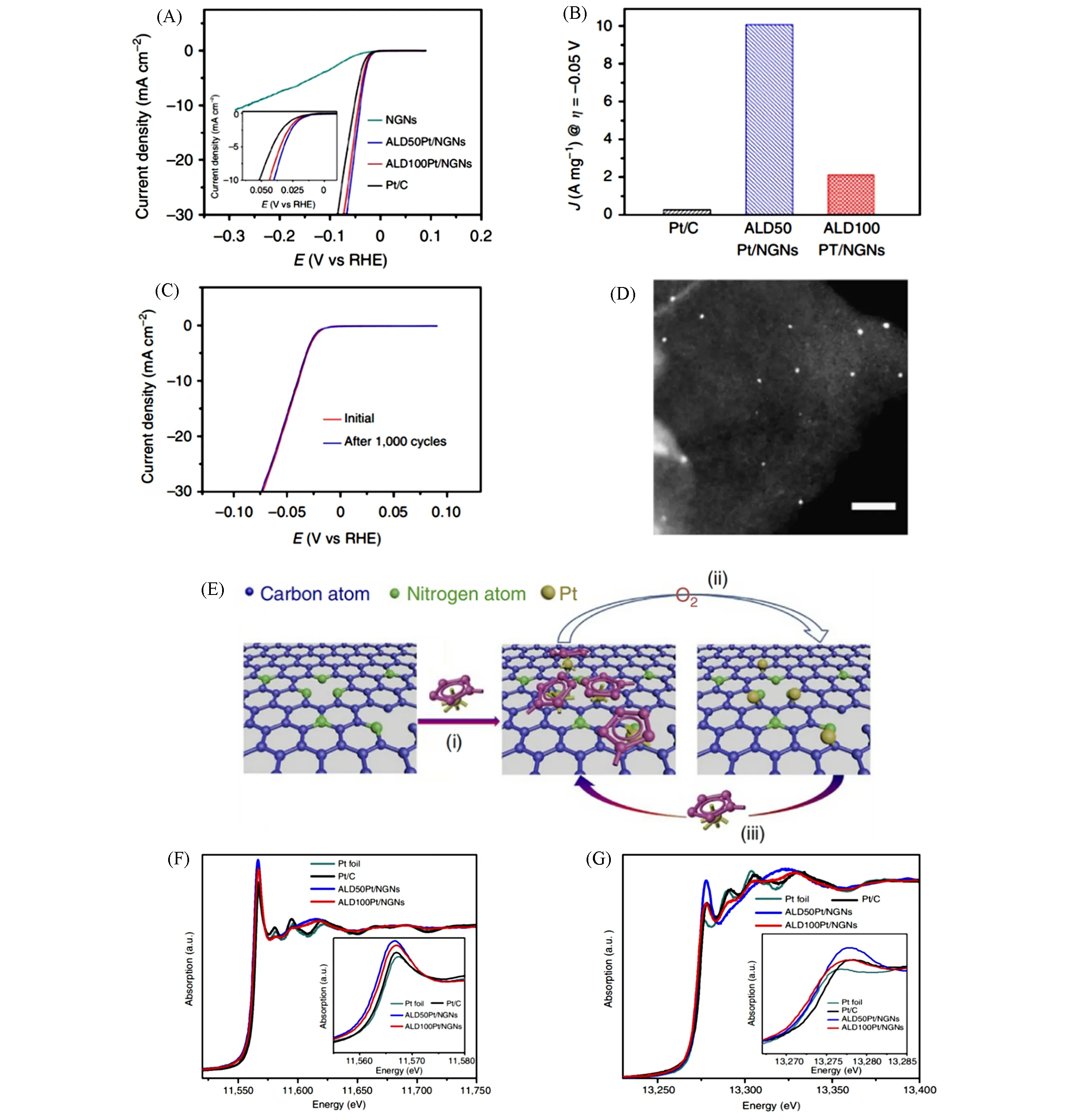
Fig.10 HER polarization curves for ALDPt/NGNs and Pt/C catalysts(A),mass activity at 0.05 V(vs.RHE)(B),durability measurement of ALD50Pt/NGNs(C),ADF⁃STEM image of ALD50Pt/NGNs(D),schematic illustration of the ALD mechanism(E),the normalized Pt L3⁃edge XANES spectra(F) and the normalized Pt L2⁃edge XANES spectra(G) of ALDPt/NGNs,Pt/C catalysts and Pt foil[77]
5 多孔材料负载Pt单原子催化剂及其同步辐射XAS
近年来,纳米限域作用被提出可以通过增强金属-载体相互作用(SMSI)来实现金属位点的高分散度,从而影响催化剂的活性和稳定性.由此衍生出的金属有机框架(MOFs)和共价有机框架(COFs)等结构受到广泛关注.考虑其在稳定金属原子和调整SMSI效应方面的独特作用,可以通过合理设计合成策略,来控制多孔材料负载金属催化剂的催化活性位点的类型和数量[78,79].
MOFs 中富含大量配位不饱和位点,并可采用配位、吸附或离子交换等方式调控金属原子的空间分布,被广泛运用于单原子催化剂的研究中[79,80].Jiang等[81]利用MOF材料作为稳定单原子的载体,并探索了其在光催化产氢方面的应用,同时对光催化过程中的电荷转移机理进行了初步探讨.MOF稳定的单原子Pt复合材料,能够在光催化裂解水产氢的过程中,使电子从MOF光敏剂转移到Pt电子受体.他们使用了一种高度稳定的铝基卟啉MOF(Al-TCPP),其通过卟啉接头将Al(OH)O4链互连成三维(3D)微孔骨架,Pt(II)吸附在卟啉中心;还原后,Al-TCPP中吡咯N原子与Pt的强相互作用可以充分锚定Pt单原子[图11(A)和(B)].结合同步辐射XANES、DRIFT和DFT计算,证明了单原子Pt表现出极高的活性,约为相同MOF稳定的Pt纳米颗粒的30倍[图11(C)~(H)].限域Pt单原子提供了高效的电子传输通道,并提高了MOF与氢的结合能,从而促进了光催化产氢[图11(I)].

Fig.11 Schematic synthesis illustration of Al⁃TCPP⁃Pt(A),aberration⁃corrected HAADF⁃STEM image of Al⁃TCPP⁃0.1Pt(B),Pt L3⁃edge XANES spectra of Al⁃TCPP⁃0.3Pt,Al⁃TCPP⁃0.1Pt,and Pt foil(C),the corresponding k3⁃weighted EXAFS spectra(D),DRIFT spectra of CO adsorbed on AlTCPP⁃0.1Pt(E),photocatalytic hydrogen production rates of Al⁃TCPP,Al⁃TCPP⁃PtNPs and Al⁃TCPP⁃0.1Pt(F),recycling performance comparison for Al⁃TCPP⁃PtNPs and Al⁃TCPP⁃0.1Pt(G),the com⁃parison of the ultrafast TA kinetics(H),the calculated free energy diagram for photocatalytic H2 production(I)[81]
2-甲基咪唑锌盐(ZIFs)也是一类特殊的金属有机框架材料;其呈四面体型3D网状结构,常作为单原子催化剂的载体[82~84].Liu等[85]用电化学还原法制备了Pt1/Co1NC催化剂;Co掺杂ZIF-8衍生的多孔碳中含有丰富的缺陷,可以提供锚定位点以捕获Pt原子并抑制团簇化[图12(A)].从HAADF-STEM和能量色散X射线光谱(EDX mapping)可以看出,Pt以原子级均匀分散在载体上[图12(B)~(D)].Pt L3边的XANES 谱中,白线(WL)峰下的区域可反映Pt5d轨道的未占状态密度;因此由WL峰的强度可见,Pt价态从高到低的顺序为PtO2>PtCl4>PtCl2>Pt1/Co1NC>Pt foil[图12(E)].进一步通过EXAFS拟合结果证明,Pt1/Co1NC 中存在Pt-N/C 共配位结构;不存在金属Pt—Pt 键,表示Pt1/Co1NC 中没有Pt 颗粒[图12(F)].Pt1/Co1NC表现出优异的析氢催化活性,过电位小于商业Pt/C催化剂[图12(G)和(H)].
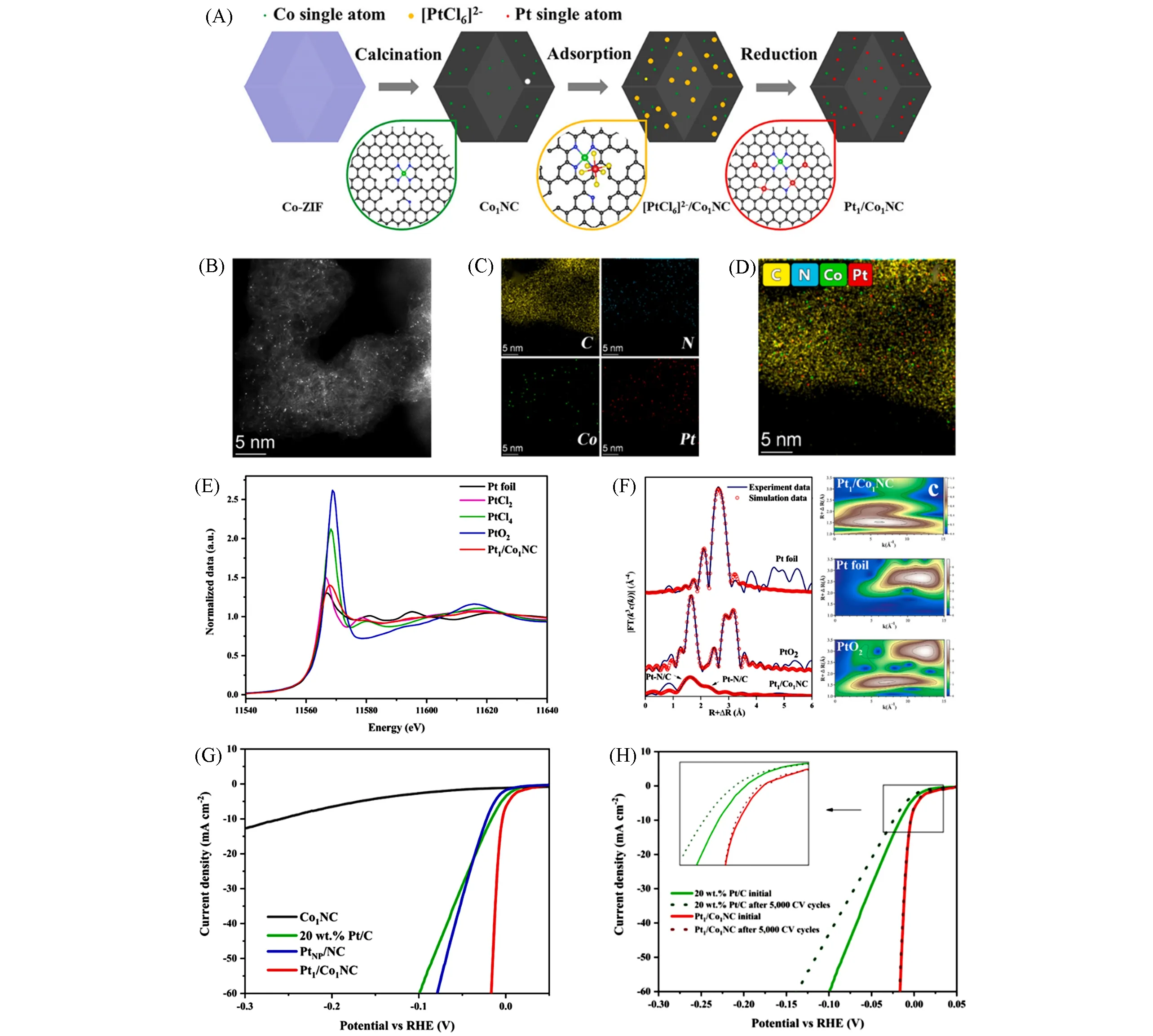
Fig.12 Schematic synthesis diagram of Pt1/Co1NC(A),HAADF⁃STEM image(B),EDX mapping images of Pt1/Co1NC(C,D),Pt L3⁃edge XANES spectra for Pt1/Co1NC and references(E),Fourier transform and wavelet transform Pt L3⁃edge EXAFS spectra(F),polarization curves of Co1NC,Pt1/Co1NC,PtNP/NC,and 20%(mass fraction)Pt/C(G),polarization curves of Pt1/Co1NC and 20%(mass fraction)Pt/C before and after 5000 CV cycles(H)[85]
6 总结与展望
随着传统化石燃料消耗量的逐年增加和世界范围内环境问题的日益严重,必须寻找清洁、可持续、可再生的新型能源以应对当前面临的挑战,因此如何制备高效、高稳定性的催化剂尤为关键.单原子催化剂的出现为解决目前的困境提供了新的思路和方法.本文综合评述了近年来金属氧化物、金属单质、纳米碳和多孔有机框架负载Pt单原子催化剂的重要研究进展,以及非原位和原位同步辐射X射线吸收谱在金属-载体相互作用解析、真实催化位点指认和构效关系理解中的应用.基于目前的研究现状,未来Pt单原子催化剂的发展及相应的同步辐射研究可以从以下几个方面展开:
(1)单原子催化剂的活性和选择性高度取决于其配位构型和电子结构以及单原子位点与载体间的相互作用,但可控调节单原子位点的配位数和局域结构极其困难.未来融合同步辐射多种技术优势的联用新方法可在指导催化剂反应微环境设计、建立结构性能关联机制方面发挥更重要的作用.
(2)尽管常规的XAS数据分析方法已被证明在研究单原子催化剂的电荷极化、局域配位和动态重构等问题上成效显著,但其对于反应吸附等变化微小的表面行为并不敏感.差分谱方法可以通过减去原位XAS测试中不变的体相信号来突出变化微小的表面信号,在探究反应吸附和催化动力学方面极具潜力[38,86,87].因此,进一步发展和完善差分谱方法对于理解铂单原子催化剂的构效关系至关重要.
(3)由于涉及反应物或中间体耦合的过程往往需要相邻的催化活性位点,因此,合成有多个金属原子的单簇催化剂是另一种提高催化效率的方法.虽然这可以通过大质量选择性、软着陆实现,但该方法成本高、产量低.从多核金属复合物前驱体中沉积是另一种选择,但其稳定性仍然是一个问题.因此,在同步辐射理解构效关系的基础上,理性设计和开发具有可控原子数的多原子催化剂未来可期.
(4)尽管目前已经提出了许多基于不同机理的Pt-SACs的合成方法,但有关大规模生产稳定、高负载SACs的报道仍然非常罕见.因此,开发简单通用的方法,来实现高负载率单原子催化剂的工业级合成及相应的大尺度同步辐射原位表征具有重要意义.
感谢合肥光源(光电子能谱站、表面催化、MCD-A和MCD-B)、上海光源(14W1,14B1)、北京光源(1W1B,4W1B,4B9A)以及中国科学技术大学微纳米研究与制造中心提供的帮助.
猜你喜欢
机电安全(2022年5期)2022-12-13
实用手外科杂志(2022年2期)2022-08-31
陶瓷学报(2021年5期)2021-11-22
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
科学(2020年1期)2020-01-06
当代陕西(2019年6期)2019-04-17
天然产物研究与开发(2018年5期)2018-06-13
中国卫生(2015年12期)2015-11-10
中国卫生(2015年10期)2015-11-10
外语学刊(2014年3期)2014-12-03
