
人发角蛋白透析纯化研究
2022-09-08 06:39郭克兵田浈桢付艳红卢伊婷季金苟郝石磊
大众科技 2022年8期
郭克兵 田浈桢 付艳红 卢伊婷 季金苟 郝石磊
人发角蛋白透析纯化研究
郭克兵1田浈桢1付艳红1卢伊婷1季金苟1郝石磊2
(1.重庆大学化学化工学院,重庆 401331;2.重庆大学生物工程学院,重庆 401331)
为了了解透析纯化过程中人发角蛋白二级结构的变化情况,用接触角测定仪、圆二色谱仪(CD)和X-射线衍射仪(XRD)等对高温氨解提取的人发角蛋白进行了分析。结果表明,透析0 h~36 h,角蛋白亲水性减小,二硫键含量增加,α-螺旋含量显著增高,无规则卷曲含量占比明显减少;透析36 h~72 h,亲水性依旧减少,二硫键含量基本不变,无规则卷曲占比显著增大。透析过程对α-螺旋和β-折叠晶区基本没有影响。
人发角蛋白;透析;纯化;时间;结构
引言
人发角蛋白因其生物相容性、良好的组织黏附性被广泛用于制作水凝胶、薄膜、支架等生物材料[1-3]。人发角蛋白通常从人发中提取而来[4,5],因此人发角蛋白的提取纯化过程受到越来越多的关注。
人发角蛋白是以α-螺旋为主要结构的α-角蛋白,属于不溶性大分子,因此难以提取[6-8]。研究者们开发了包括机械法、还原法、氧化法等方法的提取方法[4,5],但是提取过程往往包含透析纯化步骤。纯化是角蛋白应用前的必经步骤,通常透析纯化大多固定在一定时间,对其研究较小。
透析技术是最简便、最常用的分离纯化技术之一,可以去除盐、少量有机溶剂和小分子杂质,并使样品浓缩和改变微量样品的缓冲系统等。用透析袋透析简单方便、廉价、可循环利用,即将生物大分子样品溶液置入袋内,浸入水或缓冲液中,样品溶液中的大分子量的生物大分子被截留在袋内,而盐和小分子等物质不断扩散透析到袋外,直到袋内、外两边的浓度达到平衡。一般认为,角蛋白在透析过程中其二级结构会发生一定的变化,但是对变化过程未有深入的了解,现代研究发生,角蛋白的二级结构对其生物应用具有重要影响,因此,本文采用高温水氨解自制的人发角蛋白,对其透析纯化过程进行相关研究。
1 材料与方法
1.1 试剂与仪器
人发(搜集于理发店);聚氧乙烯辛基苯酚醚-10(OP-10)(成都科龙化工试剂厂);氢氧化钠、23%氨水、丙烯酰胺、三羟甲基氨基甲烷(Tris)(重庆川东化工有限公司);碳酸氢钠(成都市科隆化学品有限公司);十二烷基硫酸钠(SDS)(美国Sigma公司);考马斯亮蓝R250、过硫酸铵,(上海生工生物工程股份有限公司);醋酸(国药集团化学试剂有限公司)。以上所有试剂均为分析纯。二硫代双硝基苯甲酸(DTNB),生物试剂(上海阿拉丁生化科技股份有限公司)。蛋白上样缓冲液、彩色预染marker(11-180 kDa),生物试剂(上海源叶生物科技有限公司)。实验用水为实验室自制去离子水。
GFK-10-200型水热合成反应釜(上海贝伦仪器设备有限公司);DHG-9033A型电热恒温鼓风干燥箱(沙鹰科学仪器有限公司);MD1444(8000-14000)型透析袋,上海源叶FD-IA-50型冷冻干燥机(北京博医康实验仪器);T6新世纪型紫外可见分光光度计(北京普析通用仪器有限公司);Nicolet is50型傅立叶变换红外光谱仪(赛默飞世尔科技有限公司);1658001型电泳仪设备(美国Bio-Rad公司);CHIRASCAN型圆二色光谱仪(英国Applied Photophysics);XRD(Spectris公司)。
1.2 方法
1.2.1人发角蛋白提取
按实验室已有方法提取人发角蛋白。称取一定量人发,剪至0.5 cm~1 cm,按1︰2︰2(w/v/v)的比例向烧杯中依次加入4% OP-10和5% NaOH,室温下搅拌2 h。用去离子水冲洗6次,置于60℃烘箱过夜干燥。称取2 g已预处理的人发,置于200 mL水热合成反应釜内胆内,加入8 mL 23%氨水,将反应釜放入烘箱,185℃反应30 min。加入50 mL去离子水,室温下搅拌1.5 h,四号筛网过滤,收集滤液。滤液调pH至7.4,静置后在4 ℃下12000 rpm离心20 min,合并上清液。上清液调pH至4.0~4.2,在4℃下6000 rpm离心20 min,留沉淀。角蛋白沉淀分散于1 mol·L-1碳酸氢钠中,放入透析袋(截留分子量3500 Da)中透析,透析外液为去离子水,每12 h更换一次。
取不同透析时的角蛋白,冷冻干燥,得浅黄色粉末。
1.2.2聚丙烯酰胺凝胶电泳(SDS-PAGE)
参照Kadathur等[9]方法并稍作修改。配置12%的分离胶:1.6 mL去离子水、2 mL 30%丙烯酰胺溶液、1.3 mL 1.5 mol·L-1Tris(pH8.8)、0.05 mL 10% SDS溶液、0.05 mL 10%过硫酸铵和0.002 mL四甲基乙二胺。5%浓缩胶:2.1 mL去离子水、0.5 mL 30%丙烯酰胺混合溶液、0.38 mL 1.0 mol·L-1Tris(pH6.8)、0.03 mL 10% SDS溶液、0.03 mL 10%过硫酸铵和0.003 mL四甲基乙二胺。取20 µL角蛋白样品(15 mg·mL-1)加入5×上样缓冲液,煮沸10 min。取煮沸冷却后的样品10 µL和Marker 5 µL,上样。80 V电压下进行电泳,待Marker条带出现后,增大电压至110 V,直至电泳分离结束。考马斯亮蓝R250染色,乙醇-醋酸脱色至无背景色。
1.2.3傅里叶红外光谱(FT-IR)
将溴化钾在红外灯下于研钵中研磨成均匀粉末,再将角蛋白样品与溴化钾以1∶100的比例混合均匀后,研磨成粉。压片机20 MPa压制1 min~2 min成透明薄片,用FT-IR在400 cm-1~4000 cm-1扫描。
1.2.4接触角
将角蛋白样品压片,置于接触角测试仪平台的玻璃片上,缓慢转动旋钮,将水滴滴在在样品上,记录结果。
1.2.5二硫键含量的测定
参照Zhang等[10]方法。将角蛋白样品各称量10 mg。游离巯基含量测定:角蛋白样品加入0.8 mL缓冲液A,室温下震荡2 h,加入0.2 mL缓冲液B,继续震荡1 h,在4 ℃下10000 rpm离心10 min。上清液稀释至合适浓度,测量在412 nm的吸光度。空白参比为0.8 mL缓冲液A+0.2 mL缓冲液B。总巯基含量测定:参照游离巯基的测定方法。用缓冲液C和D替代缓冲液A和B。上述反应在避光条件下进行。计算时,摩尔消光系数取13600 M-1·cm-1,游离巯基或总巯基含量计算公式为:

式中:A412为412 nm下角蛋白的吸光度值。二硫键含量计算公式为:
二硫键含量 =(总巯基含量-游离巯基含量)/2
1.2.6圆二色谱(CD)
参照Kadathur等[11]方法。用圆二色分光光度计对浓度为0.5 mg·mL-1的角蛋白样品进行圆二色性分析。25℃,以1 nm的光谱分辨率收集数据,平均时间为1 s,扫描速率为100 nm·min-1,扫描范围190 nm~250 nm,每个样品重复测量三次。使用BeStSel服务器(http://bestsel.elte.hu)计算二级结构。
1.2.7粉末X射线衍射(XRD)
取适量角蛋白样品,研磨成细粉,过200目筛,进行粉末X射线衍射测试。
1.3 数据处理
使用GraphPad软件(Insight Science公司)进行统计分析。显著性水平设为α=0.05;用最小显著性差异法检验数据间的差异显著性,用<0.05表示。
2 结果与讨论
2.1 提取的人发角蛋白表征
2.1.1聚丙烯酰胺凝胶电泳(SDS-PAGE)
取透析了36 h所得的人发角蛋白,按照2.2.2方法,用SDS-PAGE测定其质分子量,见图1。
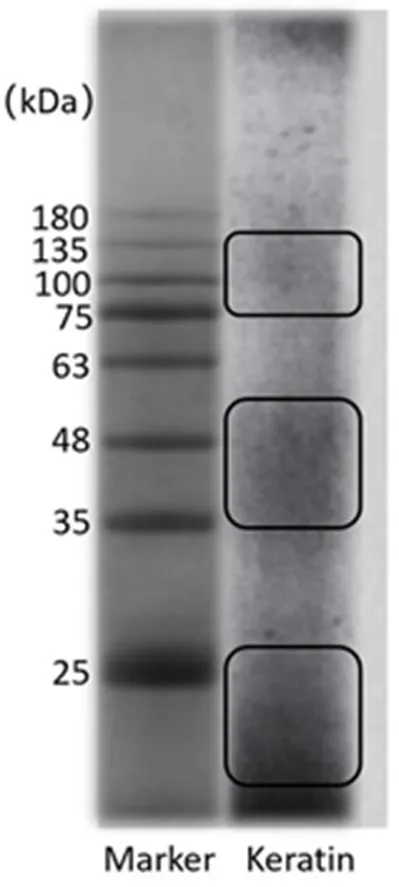
图1 角蛋白的聚丙烯酰胺凝胶电泳图
从图1可以看出,提取所得角蛋白分布在25 kDa以下、35 kDa~56 kDa、75 kDa~135 kDa,分子量分布广泛,与文献基本一致[11-13]。
2.1.2傅里叶红外光谱(FT-IR)
取透析了36 h所得的人发角蛋白,进行FT-IR分析,并和人发纤维相比较,见图2。
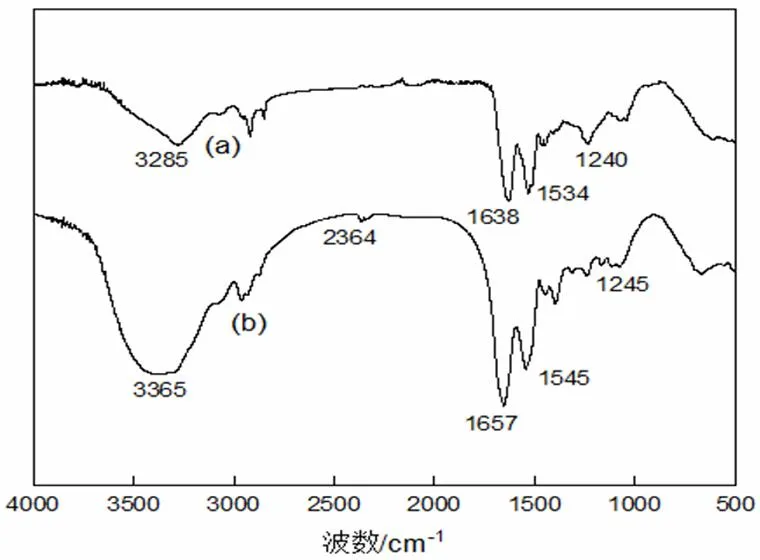
a.人发纤维;b.角蛋白
从图2可以看出,天然人发纤维与提取的人发角蛋白在3200 cm-1~3400 cm-1均具有肽键酰胺A带,1638 cm-1~1657 cm-1的酰胺I带,1534 cm-1~1545 cm-1的酰胺II带,1240 cm-1~1245 cm-1的酰胺III带,说明两者均具有典型的蛋白质结构[14,15],提取所得角蛋白与天然人发纤维分子结构相同。
综上可见,本文采用高温水氨解自制的人发角蛋白,分子量分布为25 kDa以下、35 kDa~56 kDa、75 kDa~135 kDa,具有典型的角蛋白结构。
2.2 透析时间对角蛋白的影响
角蛋白结构是影响角蛋白应用的重要因素[16]。因此,采用提取所得角蛋白,透析不同时间,研究对接触角、二硫键和分子二级结构的影响。
2.2.1接触角
为了解角蛋白亲水性的变化,采用接触角测量来直观定量。

图3 角蛋白和人皮肤的接触角图
从图3可以看出,透析时间的增加会使接触角增大,角蛋白亲水性降低。一般认为,蛋白质亲水性的好坏主要取决于分子的表面结构,如果分子表面带电氨基酸多,则亲水性高,如果疏水残基增多,则亲水性变差。之所以会发生随着透析时间增长,亲水性变差,可能是因为随着透析时间增加,一些吸水性盐被透析,或分子表面亲水残基内卷。对水的亲和能力极大地反映了角蛋白的应用前景,正常皮肤的水接触角为51.4°左右,过高或过低都不有利于其在生物修复材料中的应用,36 h和72 h角蛋白的水接触角都与正常皮肤的接触角相差不大。
2.2.2二硫键
因角蛋白的提取过程往往涉及二硫键的断裂,且二硫键在角蛋白结构中起着重要作用,所以这里测量了二硫键含量变化。
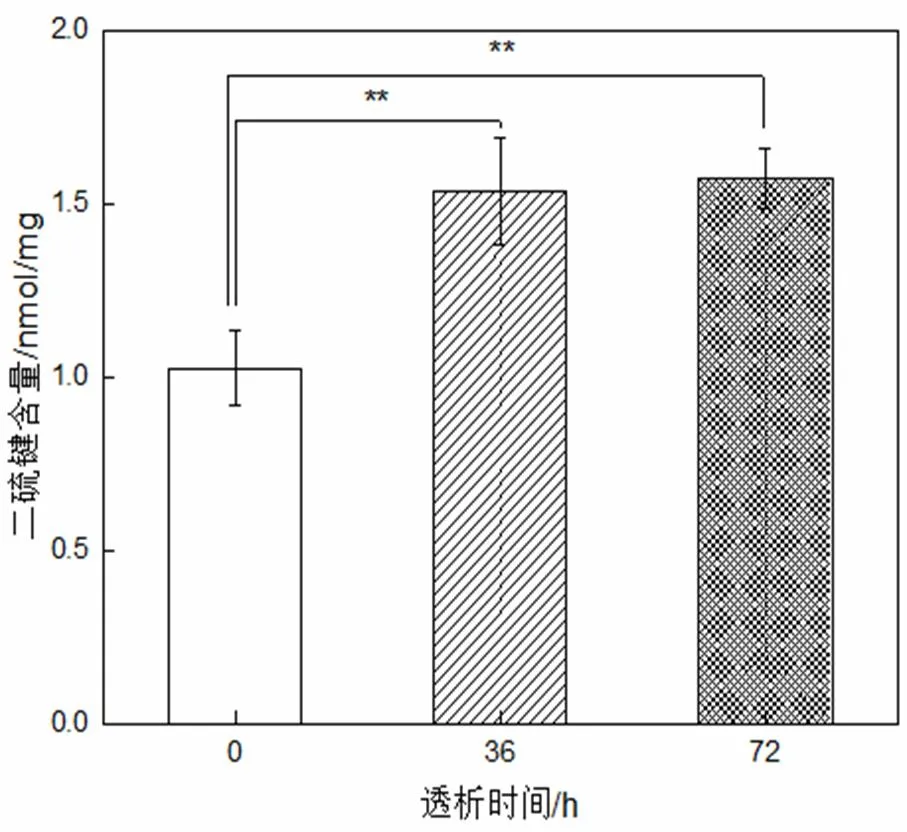
图4 角蛋白的二硫键含量
从图4可以看出,0 h~36 h,二硫键含量显著增高;36 h~72 h,二硫键含量基本不变。这可能是因为角蛋白提取涉及二硫键的断裂,初始时未含二硫键的小分子量角蛋白较多,被透析至透析外液,36 h后几乎透析干净,故36 h~72 h二硫键含量基本不变。这也提示,高温水氨解提取的人发角蛋白,透析提纯可取36 h即可,而不用透析纯化过长时间[17-19]。
2.2.3二级结构的影响
为了进一步了解透析时间对角蛋白结构的影响,用圆二色光谱(CD)对不同透析时间的角蛋白的二级结构进行了分析[21],见图5。
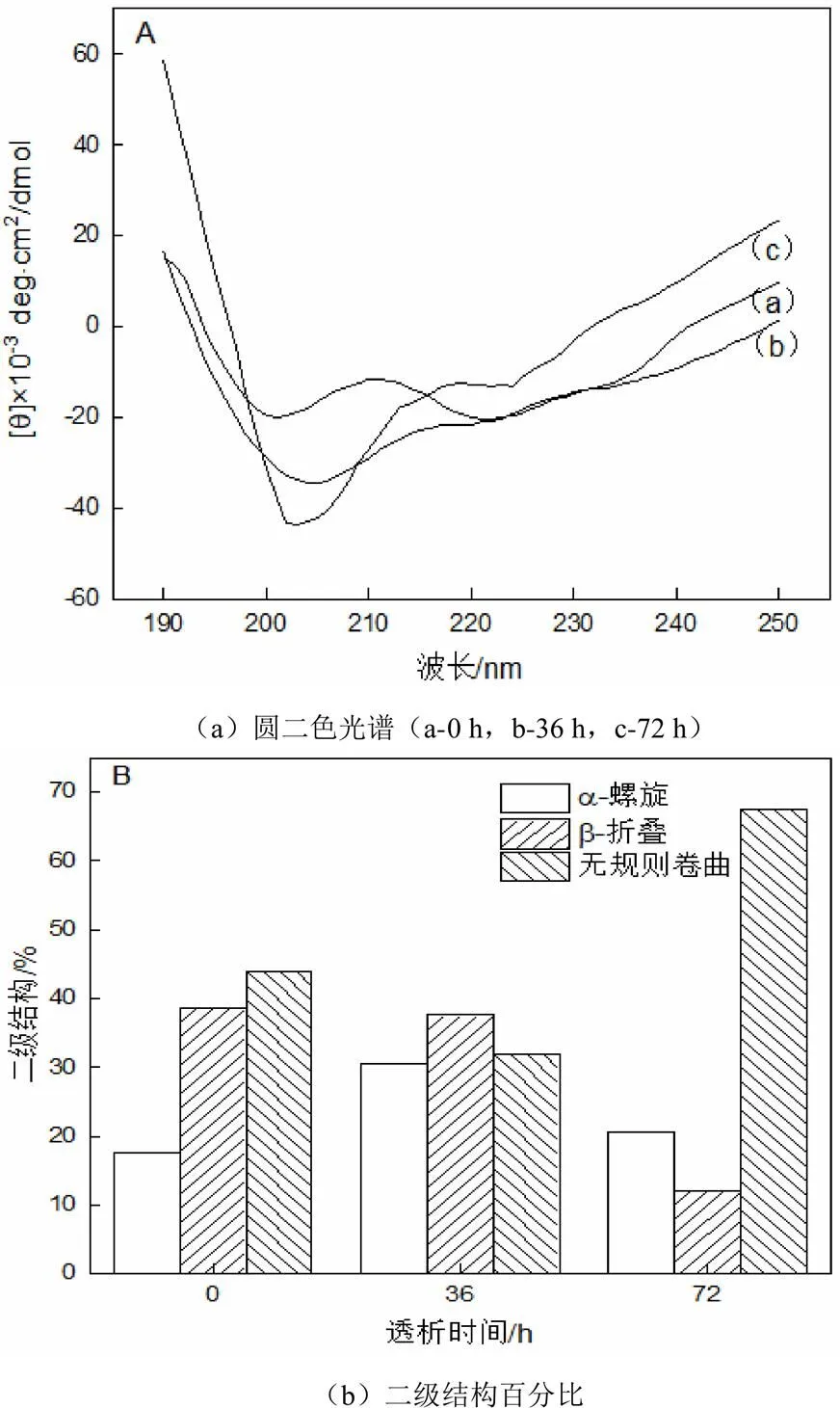
图5 不同透析时间角蛋白的二级结构
蛋白质空间结构预测的第一步通常为预测二级结构,即判断每一个氨基酸残基是否处于α-螺旋、β-折叠和或其它状态。每一段相邻的氨基酸残基具有形成一定二级结构的倾向。从图5(A)可以看出,不同角蛋白样品均在200 nm~210 nm有一个负峰,可能是α-螺旋与无规则卷曲的复合峰;在222 nm有一个负峰,属于α-螺旋。使用BeStSel服务器(http://bestsel.elte.hu)得到图5(B),可以看出,在0 h时,α-螺旋、β-折叠、无规则卷曲各占17.5%、38.6%、43.9%;在36 h时,α-螺旋、β-折叠、无规则卷曲各占31.2%、37.6%、31.2%;在72 h时,α-螺旋、β-折叠、无规则卷曲各占20.5%、12.0%、67.5%。
0~36 h透析过程中,α-螺旋含量增加,无规则卷曲减少,β-折叠含量几乎未变,这可能是因为前期透析过程中,无规则卷曲程度高的低分子量角蛋白扩散至透析外液,使无规则卷曲减少,且α-螺旋结构可能比β-折叠更稳定,故α-螺旋含量显著增高。
36 h~72 h透析过程中,无规则卷曲含量大幅度增加,可能说明在透析液中,无规则卷曲稳定性最大,由于α-螺旋相比β-折叠又更稳定,故在72 h后,无规则卷曲占比最大,α-螺旋次之,β-折叠最少。角蛋白的α-螺旋和β-折叠在长时间透析中有向无规则卷曲靠拢的趋势。结合图3说明,无规则卷曲可能主要通过内部的亲水性基团相互作用,从而使角蛋白表面疏水性增大,也变得更加稳定。
2.2.4晶型的影响
图6为角蛋白透析0 h、36 h、72 h和人发纤维的XRD图谱。
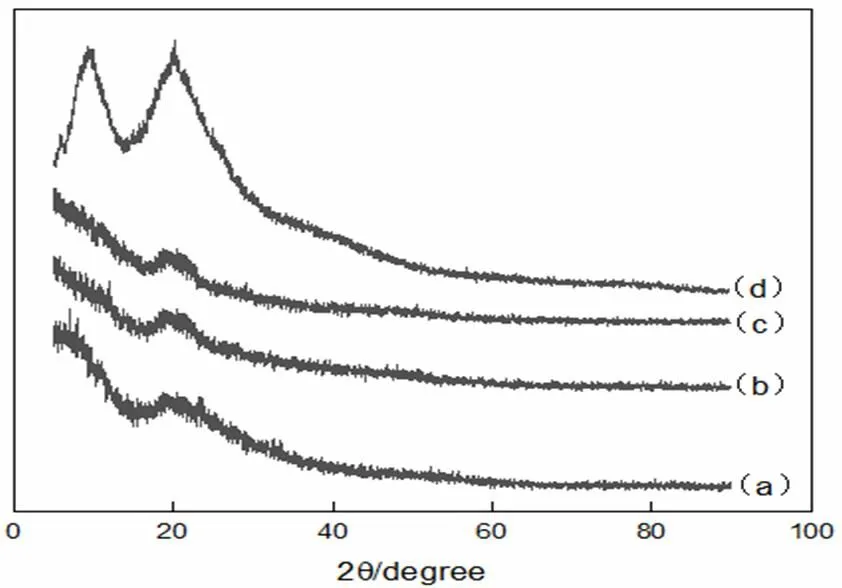
透析时间:a-0 h,b-36 h,c-72 h,d-人发纤维
在角蛋白的XRD图谱中,9°和20°左右会存在两个特征峰,9°的峰归属于α-螺旋和β-折叠,20°左右的峰则归属于β-折叠[20]。角蛋白提取物在9°和20°左右对应的峰的尖锐程度都远低于人发纤维,表明晶体化程度低。从透析0 h,36 h和72 h的XRD图可以看出,0 h和36 h的略有差别,主要表现在9°的峰形上,这可能是由于透析前的部分杂质所引起;而36 h和72 h的XRD图几乎完全一致,结合图5说明,α-螺旋或β-折叠向无规则卷曲变化过程中,α-螺旋和β-折叠晶区仍然会保留。
3 结论
本文提取人发角蛋白后,分别透析纯化0 h、36 h、72 h。结果表明,随着透析时间增加,角蛋白疏水性和二硫键均增加,透析至36 h后基本不变;随着透析的进行,α-螺旋和β-折叠,尤其是β-折叠易向无规则卷曲转变;XRD结果显示,这一转变,对α-螺旋和β-折叠微晶区基本没有影响。了解和掌握角蛋白二级结构的变化情况,对角蛋白的生物应用具有一定的指导意义。
[1] 袁小晶,尹海梦,樊晓玮,等. 角蛋白/海藻酸钠/聚丙烯酰胺水凝胶皮肤敷料的制备及创口修复研究[J]. 中国生物工程杂志,2021,41(8): 17-24.
[2] 王华杰,孙元元,卢亚楠,等. 肝素/人发角蛋白层层自组装薄膜的细胞相容性及抗凝血性研究[C]. 河南省化学会2010年学术年会论文摘要集,2010.
[3] 窦洁. 基于一氧化氮释放的角蛋白组织工程血管支架的制备研究[D]. 南京: 南京师范大学,2021.
[4] Soodeh S, Jhamak N, Azadeh G, et al. Carboxymethyl cellulose-human hair keratin hydrogel with controlled clindamycin release as antibacterial wound dressing[J]. International Journal of Biological Macromolecules, 2020, 147: 1239-1247.
[5] Fereshteh A, Mohammad T Y, Abdolhadi F, et al. Keratin nanoparticles obtained from human hair for removal of crystal violet from aqueous solution: optimized by Taguchi method[J]. International Journal of Biological Macromolecules, 2020, 143: 492-500.
[6] Chen Y, Wang Y X. Keratin and its extraction[J]. Material Review, 2002, 16(12): 65-67.
[7] Azmi N A, Idris A, Yusof N S M. Ultrasonic technology for value added products from feather keratin[J]. Ultrasonics Sonochemistry, 2018, 47: 99-107.
[8] Cassoni A C, Freixo R, Pintado A I E, et al. Novel eco-fiendly method to extract keratin from hair[J]. ACS Sustainable Chemistry and Engineering, 2018, 6(9): 12268-12274.
[9] Kadathur R R, Ramar T, Balaraman M. Comparative analysis of the chemical treatments used in keratin extraction from red sheep's hair and the cell viability evaluations of this keratin for tissue engineering applications[J]. Process Biochemistry, 2020, 90: 223-232.
[10] Zhang Z, Nie Y, Zhang Q, et al. Quantitative change in disulfide bonds and microstructure variation of regenerated wool keratin from various ionic liquids[J]. ACS Sustainable Chemistry and Engineering, 2017, 5(3): 2614-2622.
[11] Jiang X U, Ping Z, Lin Z, et al. Study of keratin extraction from human hair[J]. Wool Textile Journal, 2015, 43(5): 38-42.
[12] Idris A, Vijayaraghavan R, Rana U A, et al. Dissolution of feather keratin in ionic liquids[J]. Green chemistry, 2013, 15(2): 525-534.
[13] Xu H, Yang Y. Controlled de-cross-linking and disentanglement of feather keratin for fiber preparation via a novel process[J]. Acs Sustainable Chemistry and Engineering, 2015, 2(6): 1404-1410.
[14] Aluigi A, Tonetti C, Rombaldoni F, et al. Keratins extracted from Merino wool and Brown Alpaca fibres as potential fillers for PLLA-based biocomposites[J]. Journal of Materials Science, 2014, 49(18): 6257-6269.
[15] Wang Y X, Cao X J. Extracting keratin from chicken feathers by using a hydrophobic ionic liquid[J]. Process Biochemistry, 2012, 47(5): 896-899.
[16] 钱迅南. 丝素蛋白/角蛋白复合材料的制备与性能研究[D]. 杭州: 浙江理工大学,2020.
[17] 陈晓良. 人发角蛋白水凝胶对大鼠皮肤放射性复合伤修复促进作用的研究[D]. 重庆: 重庆医科大学,2020.
[18] 徐江涛,朱平,张林,等. 人发角蛋白提取工艺研究[J].毛纺科技,2015,43(5): 38-42.
[19] Kelly S M, Jess T J, Price N C. How to study proteins by circular dichroism[J]. Biochimica et BiophysicaActa (BBA)-Proteins and Proteomics, 2005, 1751(2): 119-139.
[20] Huang T, Rui Y, Lin Z, et al. Programing performance of wool keratin and silk fibroin composite materials by mesoscopic molecular network reconstruction[J]. Advanced Functional Materials, 2016, 26(48): 9032-9043.
Study on Dialysis and Purification of Human Hair Keratin
In order to understand the changes of the secondary structure of human hair keratin during dialysis and purification, the human hair keratin extracted by high temperature ammonolysis was analyzed by contact angle analyzer, circular dichroism (CD) and X-ray diffraction (XRD). The results showed that during 0 h -36 h of dialysis, the hydrophilicity of keratin decreased, and the content of disulfide bond increased. The α-helix content increased significantly, and the proportion of random curl content decreased significantly. During 36 h -72 h of dialysis, the hydrophilicity still decreased, the disulfide bond content remained basically unchanged, and the proportion of irregular curl increased significantly. The dialysis process had little effect on α-helix and β-folded crystal region.
human hair keratin; dialysis; purification; time; structure
Q518.1
A
1008-1151(2022)08-0043-04
2022-06-08
郭克兵(1996-),女,河北邢台人,重庆大学化学化工学院学生,研究方向为角蛋白提取机理。
季金苟(1962-),男,江西南昌人,重庆大学化学化工学院教授,研究方向为生物材料。
猜你喜欢
食品科学技术学报(2022年5期)2022-10-11
工业微生物(2020年3期)2020-06-30
分析化学(2019年4期)2019-05-13
天然产物研究与开发(2018年1期)2018-02-02
汽车实用技术(2017年24期)2018-01-24
中国饲料(2016年17期)2016-12-01
诗林(2016年5期)2016-10-25
国际皮肤性病学杂志(2016年1期)2016-03-09
中国造纸(2015年7期)2015-12-16
中华皮肤科杂志(2014年4期)2014-12-19
