
Gp350-anchored extracellular vesicles:promising vehicles for delivering therapeutic drugs of B cell malignancies
2022-09-06 13:50:24YunshnYnZhijinCi
,Yunshn Yn,Zhijin Ci,
aInstitute of Immunology,and Department of Orthopedics of the Second Affiliated Hospital,Zhejiang University School of Medicine,Hangzhou,China
bHenan Key Laboratory for Digestive Organ Transplantation,the First Affiliated Hospital of Zhengzhou University,Zhengzhou,China
cKey Laboratory of Clinical Cancer Pharmacology and Toxicology Research of Zhejiang Province,Affiliated Hangzhou First People’s Hospital,Zhejiang University School of Medicine,Hangzhou,China;
dZhejiang University Cancer Centre,Hangzhou,China
eDepartment of Critical Care Medicine of the Second Affiliated Hospital,Zhejiang University School of Medicine,Hangzhou,China
fDepartment of Emergency Medicine,Tongde Hospital of Zhejiang Province,Hangzhou,China
gSecond Affiliated Hospital of Zhejiang University School of Medicine,Zhejiang Provincial Key Lab of Ophthalmology,Hangzhou,China
hDepartment of Medical Oncology,The Cancer Hospital of the University of Chinese Academy of Sciences(Zhejiang Cancer Hospital),Institute of Basic Medicine and Cancer(IBMC),Chinese Academy of Sciences,Hangzhou,China.
Keywords:Extracellular vesicles Gp350 CD21 Red blood cells B cell malignancies
ABSTRACT Although chimeric antigen receptor-modified (CAR) T cell therapy has been successfully applied in the treatment of acute B lymphocytic leukemia,its effect on Burkitt lymphoma(BL) and chronic B lymphocytic leukemia (B-CLL) is unsatisfactory.Moreover,fatal side effects greatly impede CAR T cell application.Extracellular vesicles (EVs) are excellent carriers of therapeutic agents.Nevertheless,EVs mainly accumulate in the liver when administered without modification.As an envelope glycoprotein of Epstein-Barr viruses,gp350 can efficiently bind CD21 on B cells.Here,gp350 was directly anchored onto red blood cell EVs(RBC-EVs)via its transmembrane region combined with low-voltage electroporation.The results showed that gp350 could anchor to RBC-EVs with high efficiency and that the resulting gp350-anchored RBC-EVs (RBC-EVs/gp350Etp) exhibited increased targeting to CD21+ BL and B-CLL relative to RBC-EVs.After the loading of doxorubicin or fludarabine,RBC-EVs/gp350Etp had powerful cytotoxicity and therapeutic efficacy on CD21+ BL or B-CLL,respectively.Moreover,RBC-EVs/gp350Etp loaded with a drug did not exhibit any apparent systemic toxicity and specifically induced the apoptosis of tumor B cells but not normal B cells.Therefore,our findings indicate that drug-loaded RBC-EVs/gp350Etp may be adopted in the treatment of CD21+ B cell malignancies.
1.Introduction
At present,chimeric antigen receptor-modified (CAR) T cell therapy is considered advanced,and studies have shown CD19 is a perfect target for this therapy in most B cell malignancies[1,2].The therapy has attained impressive outcomes in individuals with acute B lymphocytic leukemia (B-ALL) [3].Although it exerts some effects in diffuse large B cell lymphoma[4],the effects of this therapy on Burkitt lymphoma(BL)remain unclear[5],and the function of CAR T cell therapy in chronic B lymphocytic leukemia (B-CLL) is limited [6].Dysfunctional CAR T cells and increased modulatory T cells in patients with B-CLL probably blunt the efficacy of CAR T cells [7,8].In addition,notable toxicities,such as cytokine release syndrome of CAR T cells,will threaten patients’lives[9].Therefore,the therapy of B cell malignancies,particularly BL and B-CLL,remains challenging[10,11].
Extracellular vesicles (EVs),including ectosomes,exosomes,oncosomes,and apoptotic bodies,are membranous vesicles released by various cells that can transfer messages between cells [12,13].The feasibility and advantages of EVs as drug carriers have been verified in many studies [14,15].EVs can not only carry proteins[16],noncoding RNAs [17],CRISPR-Cas9 plasmids [18] and chemotherapy drugs [19] but also protect these cargoes from degradation and thereby enhance their therapeutic efficacy.Compared with liposomes,which are extensively studied drug delivery vehicles,EVs are natural nanoparticles of cells with lower immunogenicity and cytotoxicity as well as better permeability [20,21].EVs from mesenchymal cells can carryKrassiRNA or shRNA to effectively suppress pancreatic cancer[22].Super-repressor IκB-loaded EVs improve survival in an LPS-induced septic mouse model[23],and EVs encapsulating chemotherapeutic drugs such as doxorubicin (Dox) exert excellent therapeutic effects on murine cancer without obvious toxicity[19].
Despite numerous explorations,the clinical translation of EVs as drug carriers remains challenging.For clinical treatment,the EV source is a critical issue to consider.Red blood cell EVs (RBC-EVs) are promising candidates due to their high yield and ideal safety [24].However,our previous study demonstrated that most unmodified RBC-EVs naturally accumulate in the liver,which greatly restricts their output efficiency for therapeutic drugs of diseases beyond the liver[25].Thus,endowing EVs with targeting properties would be meaningful.Epstein-Barr virus (EBV) primarily infects human B cells by interacting with the envelope glycoprotein gp350 and CD21 [26].Exosomes from EBV-transformed B cells containing gp350 can block EBV infection by selectively targeting mature B cells via the gp350-CD21 interaction [27].Gp350-containing EVs are B cell tropic and can transfer neoantigens to B cell malignancies [28].Therefore,gp350-modified EVs are probably optimized vehicles for the delivery of therapeutic drugs to B cell malignancies via CD21.
In this study,gp350 was effectively anchored onto RBCEVs via the transmembrane (TM) region through low-voltage electroporation,and the resulting gp350-anchored RBC-EVs(RBC-EVs/gp350Etp)showed stronger targeting to tumor CD21+B cells than RBC-EVs.Dox is a common and effective drug used for the treatment of BL [29],and fludarabine(FA) is a nucleoside analog of cytarabine that affects DNA synthesis and has been effectively used for CLL treatment[30].Correspondingly,after loading with Dox or FA,RBCEVs/gp350Etpshowed increased cytotoxicity to tumor CD21+B cells and significantly inhibited the progression of BL or BCLL,respectively.However,RBC-EVs/gp350Etploaded with Dox or FA did not lead to systemic toxicity.Therefore,our results demonstrate that the generated EVs are potential therapeutic reagents for CD21+BL and B-CLL.
2.Materials and methods
2.1.Mouse samples and human samples
Joint Ventures Sipper BK Experimental Animal Co.(Shanghai,China) provided male 6-8-week-old C57BL/6J and BALB/cnude mice,and the Model Animal Research Center of Nanjing University (Nanjing,Jiangsu,China) offered male NCG (NODPrkdcem26Cd52Il2rgem26Cd22/Nju) mice aged 6-8 weeks.All the mice were maintained in a pathogen-free facility and cared for in accordance with the institution guidelines,and the experiment was approved by the Animal Care and Use Committee of Zhejiang University (ZJU20210089).Fifteen healthy type O blood donors provided human blood (50 ml/donor),were informed of the usage of their blood,and signed consent forms.The local Ethical Committee and the Review Board of Zhejiang University approved the human blood collection(2021-045).
2.2.Cell lines
293T cells,the human B-ALL cell line SupB15,and the human B-CLL cell line MEC-1 were provided by the American Type Culture Collection (Manassas,VA,USA).The human chronic myeloid leukemia cell line K562 and the human BL cell line Raji were purchased from the Chinese Academy of Sciences.Luciferase-expressing MEC-1 (MEC-1-Luci) cells were established in the laboratory according to a previous publication[25],and 293T cells were kept in DMEM containing 10% fetal bovine serum (FBS) and penicillin/streptomycin at a concentration of 1%.K562,SupB15,Raji and MEC-1 cells were maintained in IMDM under the same conditions.All the cells were incubated at 37 degrees Celsius in a humidified environment with 5% CO2.A Mycoplasma Detection Kit(Lonza,Basel,Switzerland) was used to test all the cells for mycoplasma contamination,and the results were all negative.Shanghai Biowing Applied Biotechnology (Shanghai,China)authenticated these cell lines through DNA short-tandem repeat analysis.
2.3.Plasmid construction and transfection
The gp350 gene was synthesized by Sanyou Bio (Shanghai,China) and inserted into a pCDNA3.0-Flag (Invitrogen,New York,USA) or pET28a (Miaoling,Shanghai,China) plasmid using endonucleases and T4 ligase(TaKaRa,Dalian,Liaoning,China).PEI reagent (Polysciences,Pennsylvania,USA) was utilized for the transfection of 293T cells with pCDNA3.0-gp350-Flag based on the manufacturer’s guidance.
2.4.Isolation of EVs
We separated the RBCs from the white blood cells and plasma through a 10-min centrifugation at 500×g and passed the solution through a leukodepletion filter.We diluted the separated RBCs in dilution medium (20.5 g/l glucose,4.2 g/l NaCl,2 mM CaCl2) and treated the RBCs with 10 μM calcium ionophore (Sigma-Aldrich,St.Louis,MO,USA) overnight at 37 degrees Celsius.Afterward,the RBCs and cell debris were removed through a 20-min centrifugation at 600×g,a 15-min centrifugation at 1600×g,a 15-min centrifugation at 3260×g,and a 30-min centrifugation at 10 00×g at 4 degrees Celsius.For the isolation of 293T cell EVs,the culture supernatants of the cells were differentially centrifuged through a 10-min centrifugation at 300×g,a 20-min centrifugation at 2000×g,and a 30-min centrifugation at 10 000×g at 4 degrees Celsius.The solution was then passed through 0.22-μm syringe filters and subjected to a 70-min ultracentrifugation at 100 000×g at 4 degrees Celsius.The pellets were rinsed with a large amount of ice-cold PBS and centrifuged at 100 000×g and 4 degrees Celsius for an additional 70 min.The final pellets were resuspended in PBS.The EV protein concentrations were analyzed using a BCA Protein Assay Kit (Thermo Fisher Scientific).
2.5.Electron microscopy and nanoparticle analysis
We placed 5 μg EVs on 200-mesh copper grids coated with carbon at room temperature (RT) for 2 min.The excess suspension was removed using filter paper.The EVs were negatively stained with uranyl acetate at RT for 5 min,washed twice with PBS and subjected to a drying treatment.The EVs were measured under an FEI Tecnai T10 electron microscope (FEI,Hillsboro,OR,USA) running at 120 kV,and images were obtained.The EV grain distribution was assayed using a NanoSight NS500 instrument (Malvern,Malvern,Worcestershire,UK).
2.6.Western blotting
Ten micrograms EVs,cell lysate proteins or tumor tissues were separated via SDS-PAGE in 10% gels and transferred onto polyvinylidene difluoride membranes.The membranes were blocked with 5% powdered skim milk in PBST buffer,incubated with related primary antibodies overnight at 4 degrees Celsius and incubated with horseradish peroxidaseconjugated secondary antibodies for 1 h.Enhanced chemiluminescence reagents (MultiSciences,Hangzhou,Zhejiang,China) and a Tanon 4500 Gel Imaging System were utilized to detect the bands.Table S1 shows the informations of antibodies used in the assay and their corresponding dilutions.
2.7.Protein expression and purification
Purification of gp350 was performed according to a previous report [31].Briefly,the gp350-His-pET28a vector was selected for transformation into the BL21(DE3) strain ofE.coli(TsingKe Biotech,Beijing,China).The transformed cells with kanamycin resistance were placed on LB plates containing kanamycin and incubated overnight at 37 degrees Celsius.A single colony was then selected from the plate and cultured in 20 ml LB overnight.The obtained BL21 (DE3)-gp350 culture was adopted for grafting in 300 ml LB with violent shaking at 37 degrees Celsius.As soon as the OD600 reached 0.8,1 mM isopropyl-beta-D-thiogalactopyranoside(IPTG,Beyotime,Shanghai,China)was added,and the mixture was incubated at 24 degrees Celsius for 12 h to trigger protein overexpression.Protein-expressing bacteria were harvested by centrifugation and washed three times with PBS.These pellets were dissolved in lysis buffer(50 mM Tris,300 mM NaCl,0.2 mM PMSF,0.1%Triton X-100,pH 7.8) and lysed using an ultrasonic cell disruptor (power: 400 W;mode: on for 5 s and off for 5 s over a 20-min period)(#DH92-IIN,LAWSON,China).The prepared solution underwent a 20-min centrifugation at 20 000 g.We collected the supernatants and mixed them with pre-equilibrium nickel beads(Sangon,Shanghai,China)for 6 h at 4 degrees Celsius.The column was rinsed with urea/SDS buffer,and the proteins were eluted with 250 mM imidazole(Solarbio,Beijing,China).The final fractions were collected,and SDS-PAGE and Coomassie staining confirmed the purity of the protein.
2.8.SiRNA transfection
Raji cells were transfected with scrambled negative control (NC) orCd21siRNA using INTERFERin siRNA Transfection reagent (Polyplus,Beijing,China) according to the manufacturer’s instructions.The effects of gene silencing were then measured by quantitative PCR(qPCR)and flow cytometry.The siRNAs were synthesized by GenePharma.The sequences and primers used are listed in Table S2.
2.9.RBC-EV electroporation
RBC-EV electroporation was conducted with a BTX electroporator(Harvard Biosciences,Cambridge,MA,USA).To load the EVs with Dox (Haizheng Pharmaceuticals,Zhejiang,China) or FA (Haizheng Pharmaceuticals,Zhejiang,China),100 μg RBC-EVs and 100 μg Dox or 500 μg FA were lightly blended and incubated at 37 degrees Celsius for 60 min.After electroporation at 150 μF and 350 V in 2-mm electroporation cuvettes,the mixture was incubated at 37 degrees Celsius for 30 min to ensure complete recovery of the EV plasma membrane.To remove unincorporated Dox or FA,the EVs were rinsed twice with PBS before ultracentrifugation at 120 000×g for 90 min.The drug-loaded EVs were broken by ultrasonication(60 cycles of 10 s on and 10 s off,35%efficacy)and centrifuged at 120 000×g for 90 min.The Dox or FA concentration was measured utilizing a previously described LC-MS/MS approach[32,33].
2.10.Anchorage of gp350 onto RBC-EVs
To incorporate gp350 proteins onto RBC-EVs,100 μg RBC-EVs or drug-loaded RBC-EVs were incubated with 50 μg purified gp350 proteins for 60 min at 37 degrees Celsius and then electroporated at 50 V and 100 μF in 2-mm electroporation cuvettes utilizing a Gene Pulser Xcell electroporator(Bio-Rad,Hercules,CA,USA).Unbound gp350 proteins were removed with an Ultrafree 0.5 Biomax 100 K filter during a 20-min centrifugation at 4000×g (Millipore,Bedford,USA),and the remaining RBC-EVs/gp350Etpwere rinsed and collected in PBS.
2.11.Labeling and tracking of EVs
We labeled EVs with a VivoTrack 680 (Fluorescence,Beijing,China),PKH26,(Sigma-Aldrich) based on the manufacturer’s guidance.For VivoTrack 680 labeling,we mixed 100 μg RBCEVs in 100 μl PBS with 30 μM VivoTrack 680 at RT for 30 min.For PKH26 labeling,we resuspended 50 μg EVs in 100 μl diluent C,and 2 μl PKH26 or PKH67 dye solution was added to a separate volume of diluent C containing the same amount.Afterward,100 μl the EV suspension was mixed with 100 μl the dye solution for 5 min.For CFSE labeling,we incubated 50 μg EVs in 100 μl PBS with 7.5 μM CFSE at 37 degrees Celsius for 10 min.The staining was terminated by the addition of 100 μl FBS(EV-depleted,Thermo Fisher Scientific)and further incubation for 3 min.Ultimately,all the free dye was eliminated through a 90-min ultracentrifugation at 100 000×g,and the pellets were resuspended in 200 μl PBS Anin vivoimaging system(IVIS,PerkinElmer,Waltham,MA,USA)was used to image the uptake of VivoTrack 680-labeled EVs.
2.12.Flow cytometric analysis
To conduct this analysis,EVs were coated onto 4-μm-diameter aldehyde-sulfate latex beads (Thermo Fisher Scientific),as previously described [34].We incubated 10 μg EVs with 10 μl beads for 30 min at RT in PBS.The mixture then underwent blockage by incubation with 50 μl FBS (depleted of EVs)for 30 min.After two washes with PBS,the beads coated with EVs were stained with the related antibodies labeled with fluorescence for 30 min at 4 degrees Celsius.For the surface staining of cells,the cells were rinsed twice with PBS and incubated with the related fluorescent antibodies for 30 min on ice.After three washes with PBS,the beads or cells were analyzed via flow cytometry (NovoCyte flow cytometer,Agilent Biosciences,San Diego,CA,USA).A flow cytometric analysis of apoptotic cells was conducted by staining the cells utilizing an Annexin V-PI Apoptosis Detection Kit(MultiSciences) based on the manufacturer’s guidance.We analyzed the data using FlowJo software (Tree Star,Ashland,OR,USA).Table S1 provides the antibodies used in this study and detailed information.
2.13.Cell viability assay
The viability of Raji,K562,MEC-1 and SupB15 cells was measured through CCK-8 assays (TransGen,Beijing,China)based on the manufacturer’s protocol.The cells were treated with 10 μg/ml RBC-EVs,RBC-EVs/Dox,RBC-EVs/FA,RBCEVs/gp350Etp/Dox,RBC-EVs/gp350Etp/FA,Dox (0.3 or 5 μg/ml)or FA(1.7 or 10 μg/ml)for 24 h,48 h and 96 h in a 96-well plate,and after 10 μl CCK-8 reagent (TransGen,Beijing,China) was added to each well,the plates were incubated for 2 h at 37 degrees Celsius.The absorbance at 450 nm,which reflected the cell viability,was determined with a microplate reader.
2.14.Tumor models and treatments
We subcutaneously (s.c.) injected male BALB/c-nude mice with 1×107Raji or K562 cells on Day 0.The mice with tumors were randomized and intravenously(i.v.)injected with 100 μg RBC-EVs/Dox,RBC-EVs or RBC-EVs/gp350Etp/Dox or a routine dose of Dox (RD/Dox,5 mg/kg) every 3 d after the tumors reached 100 mm3.We monitored the tumor size every other day with a Vernier caliper.For development of a B-CLL mouse model,2×106MEC-1-Luci cells were injected i.v.into NCG mice,and 8 d later,the mice with tumors were randomized and injected i.v.with 100 μg RBC-EVs,RBC-EVs/FA,or RBCEVs/gp350Etp/FA or a routine dose of FA (RD/FA,25 mg/kg)every 3 d.The mice were imaged with an IVIS on a regular basis.For analyses of the tumor burden,the tumors were analyzed using Living Image version 4.4 software(Caliper Life Sciences).
2.15.Immunofluorescence
Tumors were collected after treatment for 10 d,embedded in Tissue-Tek Cryo-O.C.T.compound (Thermo Fisher Scientific)and cut to obtain 4-μm sections.The resulting small tumor pieces were fixed,stained with the indicated primary antibodies overnight at 4 degrees Celsius,and then stained with the related fluorescence-labeled secondary antibodies at RT for 30 min.The nuclei were then stained with DAPI for 20 min at RT.An Olympus FV3000 confocal microscope was utilized to observe the stained sections.Table S1 lists the antibodies used in this study and their corresponding dilutions.
2.16.TUNEL analysis
The TUNEL assay was conducted using the TUNEL detection kit (TUNEL,Beyotime) according to the instructions.Briefly,tumors were collected after treatment for 10 d,embedded in Tissue-Tek Cryo-O.C.T.compound (Thermo Fisher Scientific)and cut to obtain 4-μm sections.The slices were fixed in 4% paraformaldehyde at RT for 30 min.After two washes with PBS,the samples were treated with 0.3% Triton X-100 for 15 min.Subsequently,50 μl the TUNEL reaction mixture (5 μl TdT enzyme solution and 45 μl labeling solution) was added,and the resulting mixture was incubated for 1 h at 37 degrees Celsius in a dark humidified environment.The cells were costained with DAPI and visualized with an Olympus FV3000 confocal microscope.
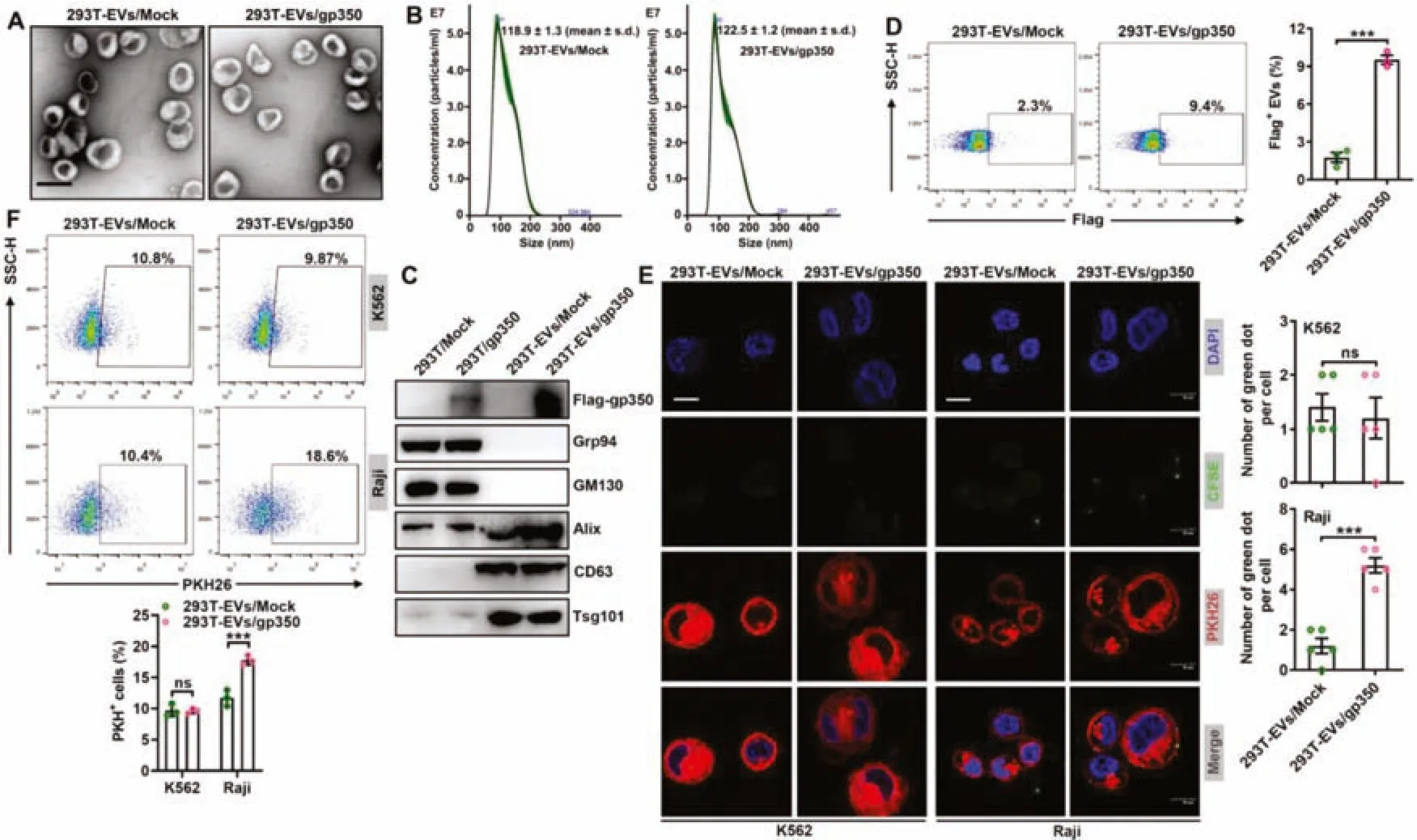
Fig.1-Modification with gp350 increases the uptake of 293T-EVs by tumor CD21+ B cells.(A-D)Mock or Flag-gp350 overexpression plasmids were transfected into 293T cells,and 48 h later,EVs were separated from the supernatants of these cells.The EV morphology was detected by TEM.Scale bar,100 nm(A).The EV grain distribution was detected using nanoparticle tracking analysis(B).The indicated proteins in the EVs were detected by immunoblotting(C).Flow cytometry was applied to analyze flag-gp350 proteins on the EVs(D).(E)PKH26-labeled K562 or Raji cells were incubated with CFSE-labeled 293T-EVs/Mock or 293T-EVs/gp350(10 μg/ml)at 4 degrees Celsius for 1 h,and the EVs on the cells were then detected by fluorescence microscopy.Scale bar,10 μm.(F)Raji or K562 cells were maintained with 10 μg/ml PKH26-labeled 293T-EVs/Mock or 293T-EVs/gp350 at 37 degrees Celsius for 1 h,and the EV uptake was then detected by flow cytometry.ns:not significant;∗∗∗P <0.001(unpaired Student’s t test).Typical outcomes from three independent trials are given(mean±SD)(n=3-5).
2.17.Histopathology
The liver,spleen,heart,kidneys and lungs from a single mouse of each of the various groups were separated after treatment for 18 d,briefly fixed in 4% paraformaldehyde and subjected to hematoxylin and eosin(H&E)staining.
2.18.Blood biochemistry
Mouse blood was collected in ethylenediaminetetraacetic acid-containing tubes (Xiangyuan Medical Apparatus Co.Ltd.,Shijiazhuang,Hebei,China) to prevent coagulation and analyzed using an ADVIA 2120 automatic hematological analyzer (Siemens,Erlangen,Germany) and an ADVIA 2400 automatic biochemistry analyzer (Siemens).The analytical measurements included a routine blood count(leukocyte and RBC levels) and assessment of the lactate dehydrogenase(LDH) and creatine kinase isoenzyme-MB (CK-MB) levels in sera.
2.19.Determination of cytokine levels
The murine serum IL-6,IL-1βand TNF levels were detected by ELISA (BioLegend,San Diego,CA,USA) based on the manufacturer’s guidance.
2.20.Isolation of CD21+ lymphocytes
Human peripheral blood mononuclear cells were isolated using lymphocyte separation medium (Haoyang bio,Tianjin,China) according to the manufacturer’s guidance.For the isolation of CD21+lymphocytes from human peripheral blood mononuclear cells,CD21+cells were further sorted with a Beckman MoFlo Astrios EQ(Beckman Coulter).
2.21.Statistical analyses
The data are expressed as the means and standard deviations(SD).The significance of the average differences between two groups was assessed using an unpaired Student’s t test,and the significance of the differences among various groups was evaluated by one-way ANOVA followed by Newman-Keuls multiple comparison test.We performed survival analyses using log-rank tests.GraphPad Prism 8.0.2(GraphPad Software Inc.,San Diego,CA,USA) software was applied for the analyses,and aP <0.05 was considered to indicate statistical significance.
3.Results and discussion
3.1.Modification with gp350 increases the uptake of 293T-EVs by tumor CD21+ B cells
To obtain EVs with membrane gp350,we transfected mock plasmids or plasmids encoding the full-lengthgp350gene,including the TM sequence,into 293T cells (termed 293T/Mock and 293T/gp350,respectively) and confirmed the overexpression of gp350 proteins (Fig.S1A).We then isolated the secreted EVs from these cells (termed 293TEVs/Mock and 293T-EVs/gp350,respectively) and confirmed that these two EVs were mainly 100 nm in diameter and had a classic two-layer membrane structure (Fig.1A).The grain distribution analysis showed that the average sizes of the 293T-EVs/Mock and 293T-EVs/gp350 were 118.9 ± 1.3 nm and 122.5 ± 1.2 nm,respectively (Fig.1B).Furthermore,both EVs were positive for the EV markers Alix,CD63 and Tsg101 and negative for the endoplasmic reticulum-residing protein GRP94 and the Golgi protein GM130,but only 293T-EVs/gp350 were positive for gp350 (Fig.1C).Flow cytometry further confirmed the existence of membrane gp350 on the surface of 293T-EVs/gp350,although at insignificant levels (Fig.1D).Subsequently,we examined whether 293T-EVs/gp350 had an increased ability to target CD21+B cells.CFSE-labeled EVs were incubated at 4 degrees Celsius with PKH26-labeled CD21+Raji cells or CD21-K562 cells(Fig.S1B).In comparison with the levels of 293T-EVs/Mock,increased levels of 293TEVs/gp350 were observed on the surface of Raji but not K562 cells (Fig.1E),which suggested enhanced binding of gp350-modified EVs to CD21+cells.Consistent with these results,we found that Raji but not K562 cells took up more PKH26-labeled 293T-EVs/gp350 than 293T-EVs/Mock during incubation at 37 degrees Celsius (Fig.1F).These data suggest that the gp350 modification effectively enhances the targeting of EVs to tumor CD21+B cells.
3.2.Successful anchorage of gp350 onto RBC-EVs via the TM region plus low-voltage electroporation
The efficiency of the loading of gp350 on 293T-EVs through the overexpression of gp350 in 293T cells was low.Moreover,as ideal EV source cells,RBCs cannot express exogenous proteins due to a lack of transcription systems [24,25].Therefore,we wanted to directly anchor gp350 onto RBC-EVs.Superantigen SEA proteins with TM domains can effectively anchor to EVs through directin vitroincubation [35].In addition,electroporation is widely used for the encapsulation of cargoes into EVs[36].Thus,we hypothesized that gp350 could be anchored on RBC-EVs through simplein vitroincubation and that electroporation might expand the spacing between membrane lipids and thereby further improve the anchoring efficiency.We confirmed that purified gp350 had the ability to more strongly bind to CD21+Raji than CD21-K562 cells(Fig.S2A).We then found that simplein vitroincubation indeed resulted in the anchorage of gp350 onto RBC-EVs(RBCEVs/gp350) (Fig.2A and 2B).In addition,the combination of incubation with low-voltage electroporation significantly enhanced the anchoring efficiency of gp350 onto RBC-EVs(RBC-EVs/gp350Etp) (Fig.2A and 2B).We then confirmed that the optimal voltage for RBC-EV electroporation was low(50 V)and that the optimal mass ratio of EVs to gp350 was 100 μg to 50 μg (Fig.S2B and S2C).After incubation with proteinase K,gp350 was no longer detected on RBC-EVs/gp350Etpby flow cytometry (Fig.2C),which further supported the membrane anchorage of gp350.In addition,gp350 could not be detected in proteinase K-treated EVs by western blotting(Fig.2D),which indicated that low-voltage electroporation did not pack gp350 into RBC-EVs.The analysis also confirmed that low-voltage electroporation did not obviously alter the morphology and grain distribution of RBC-EVs(Fig.S2D and S2E).Furthermore,both EVs were positive for the RBC marker hemoglobin A1 and the EV markers Alix and Tsg101 (Fig.S2F).To roughly evaluate the stability of gp350 anchoragein vitro,we preserved RBC-EVs/gp350Etpat 37 degrees Celsius and discovered that the anchorage was stable for at least 3 d(Fig.2E).Therefore,the combination of incubation with low voltage can directly anchor gp350 onto RBC-EVs with high efficiency.
3.3.RBC-EVs/gp350Etp exhibit enhanced targeting to tumor CD21+ B cells
We then tested the targeting of RBC-EVs/gp350Etpto tumor CD21+B cells.The amounts of RBC-EVs/gp350Etptaken up by Raji cells and CD21+MEC-1 cells (Fig.S3A) were higher than those of RBC-EVs (Fig.3A and 3B).However,the uptake of both EVs by CD21-K562 and SupB15 cells did not differ(Fig.S3B and S3C).To further confirm the role of CD21 in this process,we knocked downCd21in Raji cells and found thatCd21knockdown greatly reduced the uptake of RBCEVs/gp350Etpby Raji cells(Fig.S3D and S3E).We then assessed the CD21+tumor targeting ability of RBC-EVs/gp350Etpin vivo.Consistent with their ability to target CD21+cells,RBCEVs/gp350Etpshowed higher accumulation in Raji tumors and less accumulation in the liver than RBC-EVs (Fig.3C).However,both EVs showed similar accumulation in K562 tumors (Fig.3C).These results were further confirmed by tissue fluorescence detection(Fig.3D and S3F).To test whether gp350 anchorage could also increase the targeting of EVs to CD21+hematologic malignancies,we established CD21+MEC-1 and CD21-SupB15 leukemia cells in NCG mice.Similarly,MEC-1 cells took up more RBC-EVs/gp350Etpthan RBC-EVs,and this finding could not be obtained with SupB15 leukemia(Fig.3E).Altogether,these results suggest that the anchorage of gp350 effectively improves the CD21+tumor targeting of RBC-EVs.
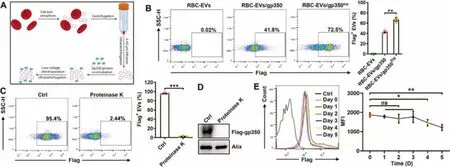
Fig.2-Successful anchorage of gp350 on RBC-EVs via the TM domain plus low-voltage electroporation.(A)Schematic diagram of the EV protein anchorage process.Briefly,100 μg of RBC-EVs was incubated with 50 μg of purified gp350 protein for 60 min at 37 degrees Celsius,and this incubation was followed by electroporation at a voltage of 50 V.(B)RBC-EVs(100 μg)were incubated with 50 μg of purified gp350 proteins for 60 min at 37 degrees Celsius(RBC-EVs/gp350),and this incubation was followed by electroporation at a voltage of 50 V(RBC-EVs/gp350Etp).Flow cytometry was employed to detect gp350 on RBC-EVs.(C,D)After treatment with 50 μg/ml proteinase K for 2 h,we measured gp350 on RBC-EVs/gp350Etp by flow cytometry(C)or western blotting(D).(E)After preservation at 37 degrees Celsius for the indicated time,gp350 on RBC-EVs/gp350Etp was analyzed by flow cytometry.MFI,mean fluorescence intensity.ns,not significant;∗P <0.05,∗∗P <0.01 and ∗∗∗P <0.001(unpaired Student’s t test).Typical outcomes from three independent trials are given(mean±SD)(n=3).
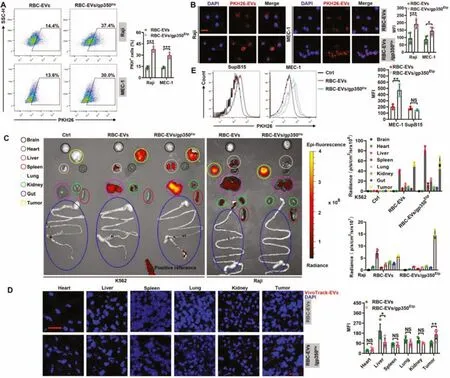
Fig.3-RBC-EVs/gp350Etp show enhanced targeting to tumor CD21+ B cells.(A,B)Raji or MEC-1 cells were maintained with 10 μg/ml PKH26-labeled RBC-EVs or RBC-EVs/gp350Etp at 37 degrees Celsius for 1 h(A)or 4 h(B).The uptake of EVs was then detected by flow cytometry(A)and fluorescence microscopy(B).Scale bar,20 μm in(B).(C,D)K562 or Raji tumor-bearing nude mice were injected i.v.with VivoTrack 680-labeled RBC-EVs or RBC-EVs/gp350Etp (100 μg)for 1 d Typical ex vivo imaging and quantification of EV uptake in the indicated organs and tumors(C).Fluorescence microscopy detection of VivoTrack 680+ cells in the indicated organ sections(D).Scale bar,20 μm in(D).(E)NCG mice were inoculated i.v.with MEC-1 or SupB15 cells for 8 d and then injected i.v.with VivoTrack 680-labeled RBC-EVs or RBC-EVs/gp350Etp (100 μg)for 1d.We then detected the MFI of MEC-1 or SupB15 tumor cells by flow cytometry analysis.ns,not significant;∗P <0.05,∗∗P <0.01 and ∗∗∗P <0.001(unpaired Student’s t test).Typical outcomes from three independent trials are given(mean±SD)(n=3-5).
3.4.Chemotherapeutic drug-loaded RBC-EVs/gp350Etp exhibit increased cytotoxicity to tumor CD21+ B cells
Since RBC-EVs/gp350Etpcould effectively target CD21+tumors,understanding whether RBC-EVs/gp350Etploaded with chemotherapeutic agents could be used to treat CD21+tumors such as BL and B-CLL is crucial.Dox and FA are common and effective drugs used for the treatment of BL and B-CLL [29,30].Thus,we loaded Dox and FA into RBC-EVs and RBC-EVs/gp350Etp(termed RBC-EVs/Dox,RBCEVs/gp350Etp/Dox,RBC-EVs/FA and RBC-EVs/gp350Etp/FA,respectively).Because Dox has autofluorescence,we first measured Dox in EVs via flow cytometry and discovered that the RBC-EVs/Dox and RBC-EVs/gp350Etp/Dox presented a similar MFI (Fig.S4A),which indicated the encapsulation of a similar amount of Dox.We also evaluated the drug loading efficiency of Dox and FA by high-performance liquid chromatography (HPLC).Based on the standard curve of Dox and FA,both RBC-EVs and RBC-EVs/gp350Etpwere loaded with similar amounts of Dox or FA (≈3 μg Dox or 17 μg FA for 100 μg RBC-EVs and RBC-EVs/gp350Etp,respectively)(Fig.S4B-S4D).Consistent with our previous study [25],the loading of the drug had no influence on the integrity of the RBC-EVs but slightly increased their size (Fig.S4E and S4F).
We next evaluated thein vitrocytotoxicity of the drugloaded RBC-EVs/gp350Etp.As expected,RBC-EVs/gp350Etp/Dox showed greater cytotoxicity in Raji cells than the same amounts of Dox (SA/Dox) and RBC-EVs/Dox,and the cytotoxicity was almost equal to that of RD/Dox at 72 h(Fig.4A).However,the RBC-EVs/gp350Etp/Dox and RBCEVs/Dox had similar inhibitory effects on K562 cells (Fig.S5A).These results indicate that the increased cytotoxicity of RBC-EVs/gp350Etp/Dox is dependent on CD21.Consistently,RBC-EVs/gp350Etp/FA had greater cytotoxic activity against MEC-1 cells than the same amounts of FA (SA/FA) and RBC-EVs/FA (Fig.4B).Moreover,a flow cytometry analysis of apoptosis revealed that RBC-EVs/gp350Etp/Dox and RBCEVs/gp350Etp/FA,but not RBC-EVs/Dox and RBC-EVs/FA,exerted more proapoptotic effects on Raji and MEC-1 cells,respectively(Fig.4C and 4D).However,RBC-EVs/gp350Etp/Dox and RBC-EVs/gp350Etp/FA had similar proapoptotic effects on K562 cells (Fig.S5B).The above-described observations demonstrate that RBC-EVs/gp350Etp/Dox and RBCEVs/gp350Etp/FA are more cytotoxic to tumor CD21+B cellsin vitro.
3.5.Chemotherapeutic drug-loaded RBC-EVs/gp350Etp exert stronger effects against BL and B-CLL
We subsequently examined the effects of RBCEVs/gp350Etp/Dox and RBC-EVs/gp350Etp/FA against BL and B-CLLin vivo.First,we determined the optimal treatment dose of EVs and found that 100 μg RBC-EVs/gp350Etp/Dox showed ideal treatment effects on Raji tumors compared with 150 μg RBC-EVs/gp350Etp/Dox (Fig.S5C);thus,a dose of 100 μg was selected for subsequent experiments.The administration of SA/Dox and RBC-EVs/Dox had no inhibitory effect on the growth of Raji tumors,and RBCEVs/gp350Etp/Dox significantly inhibited Raji tumor growth to levels almost equal to those obtained with RD/Dox treatment(Fig.5A).No difference in tumor size was found between the RBC-EVs/gp350Etp/Dox and RD/Dox treatments,but RBC-EVs/gp350Etp/Dox treatment significantly extended the survival of Raji tumor-bearing mice in compared with that obtained with RD/Dox treatment (Fig.5B).TUNEL assays of tumor tissues showed that RBC-EVs/gp350Etp/Dox treatment led to a more notable increase in Raji cell apoptosis than no treatment and RBC-EVs/Dox treatment (Fig.5C).However,this effect of RBC-EVs/gp350Etp/Dox was significantly weaker than that of RD/Dox(Fig.5C).Moreover,immunofluorescence staining of the proliferation marker Ki67 revealed that RBC-EVs/gp350Etp/Dox treatment significantly reduced the proliferation of tumor cells,and this reduction was almost equal to that obtained with RD/Dox(Fig.5C).Furthermore,we confirmed that cell death induced by RBC-EVs/gp350Etp/Dox was related to the classical apoptotic pathway,as evidenced by a reduction in Bcl2 and an increase in cleaved caspase 3 in tumor tissues (Fig.5D).The analysis of CD21-K562 tumors revealed that neither RBC-EVs/gp350Etp/Dox nor RBCEV/Dox exerted obvious inhibitory effects,whereas RD/Dox significantly inhibited the progression of these tumors (Fig.S5D).Moreover,RD/Dox but not RBC-EVs/gp350Etp/Dox and RBC-EV/Dox extended the survival time of K562 tumorbearing mice(Fig.S5E).
We then evaluated the effects of RBC-EVs/gp350Etp/FA against B-CLL by introducing MEC-1-Luci leukemia into NCG mice.More significant tumor suppression was observed in mice that received RBC-EVs/gp350Etp/FA than in those that received RBC-EVs/FA,but this suppression was weaker than that obtained with RD/FA (Fig.5E).In addition,significantly longer survival was observed among the RBC-EVs/gp350Etp/FA-and RD/FA-treated mice (Fig.5F).However,unlike the results found for the magnitude of tumor suppression,RBC-EVs/gp350Etp/FA and RD/FA had similar effects on the survival of tumor-bearing mice(Fig.5F).Collectively,the results demonstrate that Dox-or FA-loaded RBC-EVs/gp350Etpare effective in treating CD21+BL and B-CLL.

Fig.4-Chemotherapeutic drug-loaded RBC-EVs/gp350Etp exhibit increased cytotoxicity to tumor CD21+ B cells.(A)After treatment with SA/Dox(0.3 μg/ml Dox),RBC-EVs/Dox,RBC-EVs/gp350Etp/Dox,or RD/Dox(5 μg/ml Dox)for the indicated times,we measured the viability of Raji cells with CCK-8 assays.(B)After treatment with SA/FA(1.7 μg/ml FA),RBC-EVs/FA,RBC-EVs/gp350Etp/FA,or RD/FA(10 μg/ml FA)for the indicated times,the MEC-1 cell viability was measured with CCK-8 assays.(C)After treatment with SA/Dox(0.3 μg/ml Dox),RBC-EVs/Dox,RBC-EVs/gp350Etp/Dox,or RD/Dox(5 μg/ml Dox)for 24 h,we measured Raji cell apoptosis by flow cytometry utilizing an Annexin V staining kit.(D)After treatment with SA/FA(1.7 μg/ml FA),RBC-EVs/FA,RBC-EVs/gp350Etp/FA,or RD/FA(10 μg/ml FA)for 1 d,the apoptosis rate of MEC-1 cells was determined via flow cytometry utilizing an Annexin V/PI staining kit.ns,not significant;∗P <0.05;∗∗P <0.01 and ∗∗∗P <0.001(one-way ANOVA followed by Newman-Keuls multiple comparison test).Typical outcomes from three independent trials are given(mean±SD)(n=3).
3.6.Drug toxicity evaluation of RBC-EVs/gp350Etp/Dox and RBC-EVs/gp350Etp/FA
To investigate the potential clinical application of RBCEVs/gp350Etp/Dox and RBC-EVs/gp350Etp/FA,we further assessed the toxicity of RBC-EVs/gp350Etp/Dox and RBCEVs/gp350Etp/FAin vivo.RBC-EVs/gp350Etp/Dox did not cause severe weight loss,whereas RD/Dox did exert this effect(Fig.6A).Cardiotoxicity is the major side effect associated with Dox [37,38].Thus,we evaluated the CK-MB and LDH levels in sera because these compounds are marker enzymes of myocardial damage [39].Marked increases in the serum CK-MB and LDH levels were observed in the RD/Dox-treated mice but not RBC-EVs/gp350Etp/Dox-treated mice (Fig.6B).Furthermore,the RBC-EVs/gp350Etp/Dox-treated mice showed no obvious histopathological damage in the five main organs stated previously,and the same was true for the RD/Doxtreated mice with the exception of marked heart damage(Fig.6C).Another side effect of the clinical use of Dox is systemic myelosuppression,which limits intensification of its dose[40,41].As expected,the leukocyte and RBC numbers were markedly decreased in the RD/Dox-treated mice,whereas the numbers of both of these cells remained unchanged in the RBC-EVs/gp350Etp/Dox-treated mice (Fig.6D).In addition,Dox can induce the release of a vast number of inflammatory cytokines [42].Indeed,RD/Dox treatment greatly increased the serum levels of proinflammatory cytokines,including IL-1β,IL-6 and TNF,whereas RBC-EVs/gp350Etp/Dox did not obviously alter the serum levels of these cytokines (Fig.6E).FA reportedly causes myelosuppression [43],autoimmune hemolytic anemia [44] and eosinophilia [45].RD/FA but not RBC-EVs/gp350Etp/FA reduced the blood leukocyte numbers,which indicated that RD/FA induces myelosuppression (Fig.6F).However,in our model,we did not observe any significant changes in blood RBCs,hemoglobin or eosinophils in the mice administered RD/FA or RBC-EVs/gp350Etp/FA(Fig.6F).To consider their future clinical applications,we also tested the cytotoxicity of RBC-EVs/gp350Etp/FA to CD21+lymphocytes.We found that although RBC-EVs/gp350Etp/FA greatly induced the apoptosis of MEC-1 cells,they exhibited almost no cytotoxicity to CD21+lymphocytes (Fig.6G),which was probably caused by a reduced uptake of RBC-EVs/gp350Etp/FA by CD21+lymphocytes than by MEC-1 cells(Fig.S6A and S6B).Thus,these data suggest that RBC-EVs/gp350Etp/Dox and RBC-EVs/gp350Etp/FA have satisfactory biosafety.
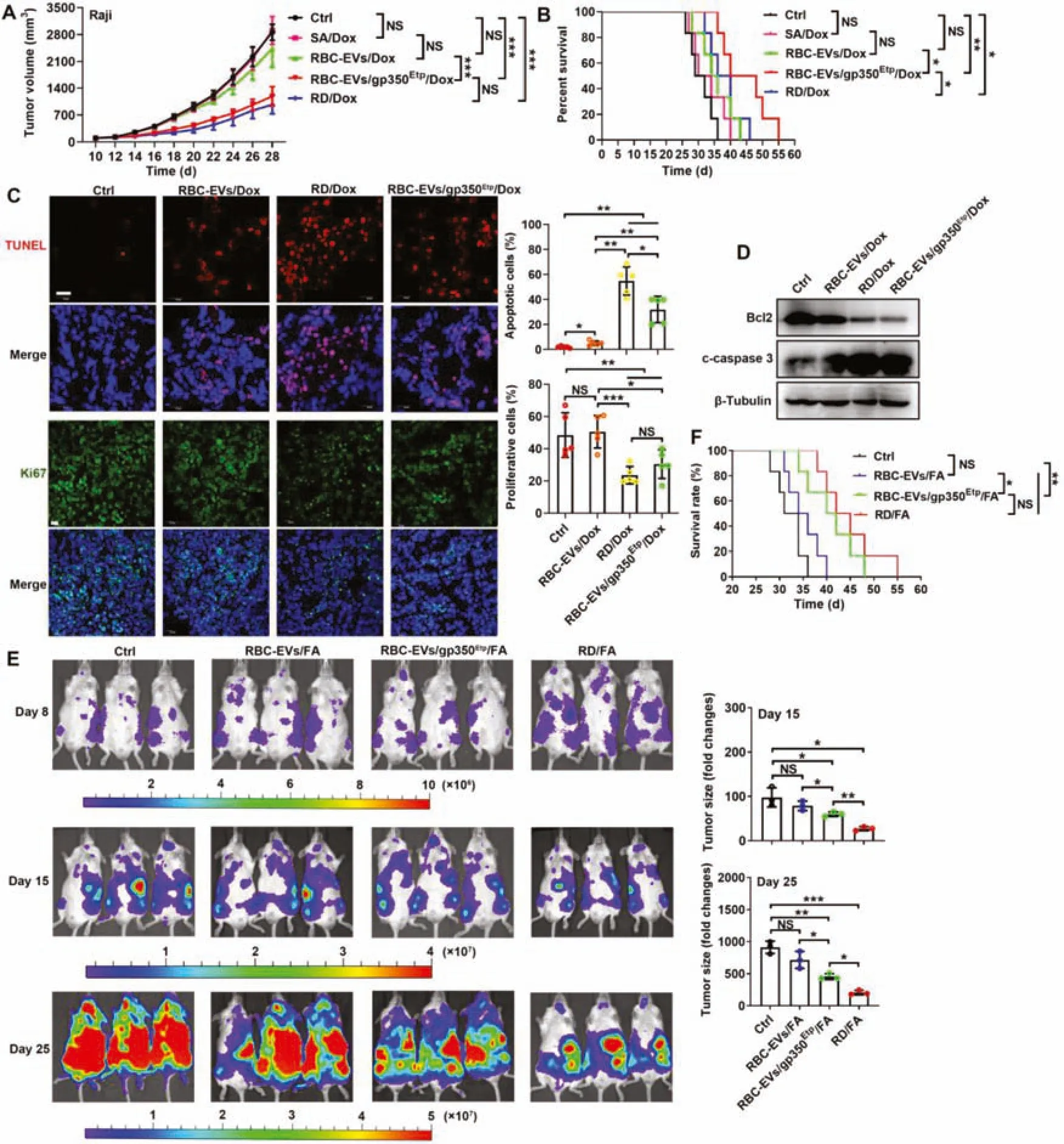
Fig.5-Chemotherapeutic drug-loaded RBC-EVs/gp350Etp exert stronger effects against BL and B-CLL.(A-D)7 d after injection with 1×107 Raji cells,mice were injected i.v.with RBC-EVs(100 μg),RBC-EVs/Dox(100 μg),SA/Dox(3 μg Dox),RD/Dox(5 mg/kg Dox)or 100 μg of RBC-EVs/gp350Etp/Dox every 3 d.Tumor progression was evaluated based on the tumor size(A)and survival time(B).Representative images of TUNEL and Ki67 immunofluorescence staining of tumor tissues from mice subjected to the indicated treatment for 10 d are shown(left)and were statistically analyzed(right).Scale bars,20 μm(C).WB analysis of Bcl2 and cleaved caspase 3(c-caspase 3)proteins in tumor tissues from mice subjected to the indicated treatment for 10 d(D).(E,F)NCG mice were injected i.v.with 2×106 MEC-1-Luci cells,and 8 d later,the mice were injected i.v.with 100 μg RBC-EVs,100 μg RBC-EVs/FA,100 μg RBC-EVs/gp350Etp/FA or RD/FA(25 mg/kg FA)every 3 d.The tumors were monitored via IVIS on Days 8,15 and 25(left).We evaluated tumor progression by computing the tumor signal intensity on Day 15 or 25 divided by that on Day 8(right)(E)and based on the survival(F).ns,not significant;∗P <0.05;∗∗P<0.01 and ∗∗∗P <0.001(one-way ANOVA followed by Newman-Keuls multiple comparison test).Typical outcomes from three independent trials are given(mean±SD)(n=3-7).
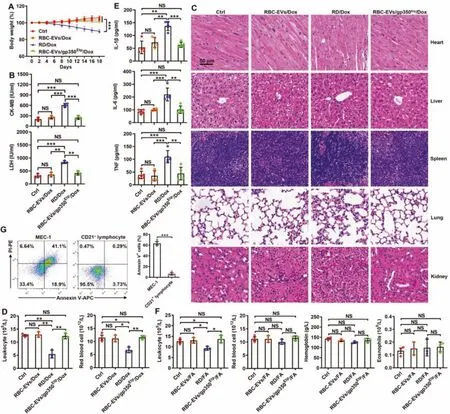
Fig.6-Drug toxicity evaluation of RBC-EVs/gp350Etp/Dox and RBC-EVs/gp350Etp/FA.(A-E)C57BL/6J mice were treated with RBC-EVs/gp350Etp/Dox(100 μg)or RD/Dox(5 mg/kg Dox)every 3 d.The body weights were recorded every other day(A).The serum LDH and CK-MB levels were measured after 10 d(B).Representative images of five major organs(H&E stained)after 18 d.Scale bar,50 μm(C).Blood leukocytes and RBCs(D)and serum inflammatory cytokines(IL-1β,IL-6 and TNF)(E)were detected after 10 d.(F)C57BL/6J mice were treated with RBC-EVs/gp350Etp/FA(100 μg)or RD/FA(25 mg/kg FA)every 3 d.The numbers of blood leukocytes,RBCs,and eosinophils and the hemoglobin levels were measured after 10 d.(G)MEC-1 cells or CD21+ lymphocytes were cultured with or without 10 μg/ml RBC-EVs/gp350Etp/FA at 37 degrees Celsius for 12 h.The apoptosis of cells was measured by flow cytometry using an Annexin V-PI staining kit.ns:not significant;∗P <0.05;∗∗P <0.01 and ∗∗∗P <0.001(one-way ANOVA followed by Newman-Keuls multiple comparison test).Typical outcomes from three independent trials are given(mean±SD)(n=3-5).
4.Conclusion
Here,we directly anchored gp350 onto RBC-EVs via its TM region and utilized the characteristic that gp350 specifically binds to CD21 on B cells to make RBC-EVs that target CD21+B cell tumors.We combined this approach with low-voltage electroporation to further improve the anchoring efficiency of gp350.After loading with Dox or FA,RBC-EVs/gp350Etpshowed powerful cytotoxicity to BL or B-CLL,respectively.Unlike CAR T cells,which cause deadly cytokine release syndrome,drugloaded RBC-EVs/gp350Etppossess excellent safety and thus show notable therapeutic effects on these malignancies.
In conclusion,this study established a new strategy to efficiently anchor gp350 onto RBC-EVs.BL or B-CLL,whose therapy remains challenging,could be effectively inhibited by gp350-anchored RBC-EVs loaded with Dox or FA,respectively.Hence,our findings will contribute to the exploration of new strategies for BL and B-CLL therapy.
Conflicts of interest
The authors report no conflicts of interest.The authors alone are responsible for the content and writing of this article.
Acknowledgements
This work was financially supported by the Natural Science Foundation of Zhejiang Province (LY19H160009 and LY20H120007),the National Natural Science Foundation of China (82130053,81971871,31970845 and 81901571),the Joint Preresearch Fund for Clinical Scientific Research of Hangzhou First People’s Hospital Affiliated to Zhejiang University (YYJJ2019Z07) and the Major Project of Hangzhou Health Science and Technology Plan(Z20200134).The authors would like to thank Zhaoxiaonan Lin (Core Facilities of Zhejiang University school of Medicine,Hangzhou,China)for her technical assistance in the immunofluorescence.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.ajps.2022.03.004.
 Asian Journal of Pharmacentical Sciences2022年3期
Asian Journal of Pharmacentical Sciences2022年3期
- Asian Journal of Pharmacentical Sciences的其它文章
- Salvianolic acid B dry powder inhaler for the treatment of idiopathic pulmonary fibrosis
- New modular platform based on multi-adjuvanted amphiphilic chitosan nanoparticles for efficient lipopeptide vaccine delivery against group A streptococcus
- Sequentially releasing self-healing hydrogel fabricated with TGFβ3-microspheres and bFGF to facilitate rat alveolar bone defect repair
- Elaborately engineering of a dual-drug co-assembled nanomedicine for boosting immunogenic cell death and enhancing triple negative breast cancer treatment
- Changes in target ability of nanoparticles due to protein corona composition and disease state
- Exosomes-mediated tumor treatment:One body plays multiple roles