
液质联用分析测定硝基呫吨酮的方法改进研究
2022-09-02 09:18于保青
世界农药 2022年8期
于保青
(上海晓明检测技术服务有限公司理化分析室,上海 201612)
硝磺草酮是由先正达公司于2001年研发上市的三酮类除草剂,其通过抑制杂草的羟基苯基丙酮酸酯双氧化酶(HPPD)的活性而杀灭杂草,应用于玉米、水稻、甘蔗等作物防除阔叶和禾本科杂草。硝磺草酮高效、杀草谱广,是当前应用非常广泛的一个除草剂品种[1]。硝基呫吨酮(图1)是合成硝磺草酮过程中可能产生的1个杂质,因该物质的Ames试验结果为阳性,其对人类的健康有潜在的风险,所 以EFSA将其定为相关杂质,规定在硝磺草酮原药中限量最大值为2 mg/kg[2]。
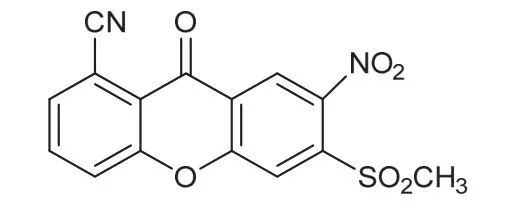
图1 硝基呫吨酮的化学结构式
关于硝磺草酮原药中的硝基呫吨酮定量分析,王英等开发了HPLC分析方法,其最低定量限(LOQ)较高为1.5 mg/kg,且该方法对基线要求较高[3]。李东等开发的LCMS外标定量法,根据其标准曲线计算,定量范围约为2~50 mg/kg,LOQ等于EFSA规定的限量值[4]。硝磺草酮原药的国家标准(编号:GB29382—2012)[5]中描述的硝基呫吨酮用LCMS单点外标法进行定量分析,方法中描述的标准溶液浓度为10 mg/kg,浓度高于EFSA规定的限量值,且如果样品溶液中浓度与标准溶液浓度相比差别较大时,则不能保证定量的准确性。为确保产品生产质量稳定合格,生产企业在监测生产工艺的稳定性时,往往需要对远低于EFSA规定的限量值的浓度进行定量。
根据生产企业的需要,本研究对国家标准GB 29382—2012中的硝基呫吨酮的分析方法进行了改进,包括离子源类型、扫描模式、扫描离子对、流动相中盐的浓度、流速和定量方法,以期提高方法的灵敏度、扩大定量范围、提高分析效率,为生产企业检测该杂质提供方法参考。
1 试验部分
1.1 仪器和试剂
Secix QTRAP 4500液质联用仪(美国AB Secix公司);MS205DU电子分析天平(0.000 1 g,梅特勒-托利多);BT25S电子分析天平(0.000 01 g,赛多利斯)和SK3300H超声波清洗器(上海科导超声仪器有限公司);Milli-Q Direct8纯水-超纯水一体机(美国密理博公司)。
乙腈(色谱纯,美国TEDIA公司);乙酸铵(色谱纯,上海麦克林生化科技有限公司);超纯水(纯水-超纯水一体机自制)。
硝基呫吨酮标样:含量为99.5%,购于国家农药质检中心(沈阳);硝磺草酮原药样品A:不含硝基呫吨酮,由上海泰禾化工有限公司技术研发中心提供;硝磺草酮原药样品B:含硝基呫吨酮,由上海泰禾化工有限公司技术研发中心提供。
1.2 样品制备
1.2.1 标准溶液的制备
称取0.005 g (精确至0.000 01 g)硝基呫吨酮标样于25 mL容量瓶中,加入少量乙腈后超声,使标样充分溶解,冷却至室温,用乙腈定容至刻度,摇匀,命名为储备液A;移取0.25 mL储备液A至50 mL容量瓶,用乙腈稀释定容得浓度为1 mg/L溶液,命名为储备液B。用乙腈逐级稀释储备液B,得到浓度为0.005、0.01、0.02、0.05、0.1、0.5 mg/L的硝基呫吨酮溶液。
1.2.2 样品溶液的制备
称取硝磺草酮样品约0.5 g (精确到0.000 01 g)于10 mL刻度试管内,加入适量乙腈溶解并稀释至5 mL,混匀,过微孔滤膜待用。
1.3 仪器分析条件
1.3.1 液相色谱条件
液相色谱柱:GL Sciences Inert Sustain 150 mm× 4.6 mm,5 μm;柱温:40 ℃;流速1.0 mL/min;进样体积:5.0 μL;流动相:乙腈:1 mM/L乙酸铵水溶液=50∶50 (体积比);运行时间:6 min;硝基呫吨酮保留时间约为4.8 min。典型色谱图见图2。
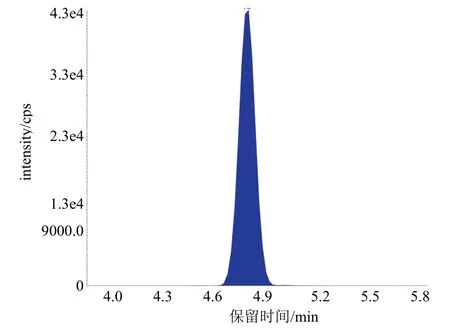
图2 硝基呫吨酮的MRM (345.0/235.1)提取离子色谱图
1.3.2 质谱条件
电离源模式:ESI离子源;扫描方式:MRM 正离子;离子源电压:5 500 V;离子源温度:500 ℃;Curtain Gas:40 Psi;Gas 1:50 Psi;Gas 2:50 Psi;转换阀:0~4 min(进入废液),4~6 min(进入质谱)。
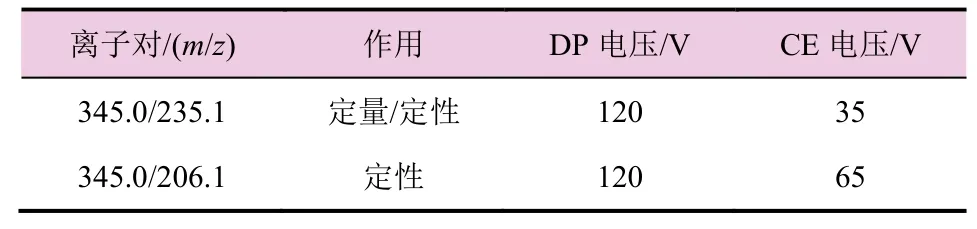
表1 质谱参数
1.4 测定
在上述1.3节操作条件下,待仪器稳定后先进一针空白溶剂(乙腈),再按照浓度从低到高的顺序测定标样溶液,然后进一针或更多空白溶剂(乙腈)以检查液相及质谱体系内有无残留干扰,而后进空白样品(样品A),最后测定精密度样品溶液以及回收率样品溶液。
1.5 计算
以标准溶液的浓度为横坐标,峰面积为纵坐标,用最小二乘法进行线性回归,得到回归方程和相关系数。
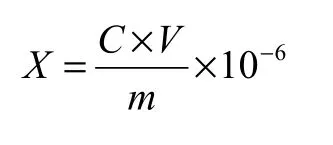
将精密度样品溶液以及回收率样品溶液的峰面积代入回归方程,计算得到样品的硝基呫吨酮浓度。试样中硝基呫吨酮的质量分数按下式计算: 式中:X为硝基呫吨酮的质量分数,mg/kg;C为回收率/精密度样品溶液中硝基呫吨酮的浓度,mg/L;V为回收率/精密度样品溶液的体积,mL;m为回收率/精密度样品的称样量,g;10-6为单位转换系数。
2 结果与讨论
2.1 仪器操作条件的改进
在国标GB29382—2012方法基础上,本研究进行了以下条件改进:⑴ ESI源代替了APCI源,因为对于液质联用仪来说,ESI源比APCI源使用频度更高,更通用,可减少离子源的更换操作。⑵ 用正离子扫描模式代替国标中的负离子扫描模式,因为硝基呫吨酮在正离子模式下信号响应明显高于负离子模式。试验结果显示,本方法的LOQ为0.1 mg/kg,与文献的1.5 mg/kg相比,灵敏度提高了15倍比。⑶ 选择了2个扫描离子对,1个用于定量,2个用于定性。与国标方法只有1个离子对进行定性定量相比,2个离子定性可更多地排除干扰物质,使方法的特异性更好。⑷ 将国标规定的流动相中乙酸铵水溶液浓度由5 mM/L下调到1 mM/L,原因是流动相中的乙酸铵有助于硝基呫吨酮与硝磺草酮在色谱柱上进行很好的分离,从而将样品中大量的硝磺草酮切到废液中而避免其进入质谱产生系统污染的风险。但是流动相溶液中较高浓度的乙酸铵在正离子模式下对信号会有显著的抑制作用。在综合考虑下,本方法采用乙酸铵水溶液的浓度为1 mM/L。⑸ 将国标中的0.5 mL/min流速调到1.0 mL/min。流速提高有助于缩短分析时间。国标中的分析时间为10 min,目标物出峰时间为7.2 min;本研究的分析时间为6 min,目标物出峰时间为4.8 min。分析时间节省了4 min。⑹ 采用标准曲线外标定量法代替国标中的单点外标定量。考虑到标准曲线的截距通常不为零,一般样品溶液与标样溶液的浓度相比偏差超出±20%时,单点外标定量法不再适应。本研究标准曲线的高低浓度范围是200倍,比单点的范围更宽。当样品中目标物的浓度未知时,采用标准曲线定量更适合。
2.2 专属性试验
在1.3节分析条件下,分别进样溶剂(乙腈)、空白样品(样品A)、硝基呫吨酮标样和样品B,以检查在目标峰保留时间及其附近是否有干扰峰出现。结果以上样品在目标峰的保留时间及其附近均无干扰峰出现。
2.3 线性试验
以溶液质量浓度为横坐标,以色谱峰面积为纵坐标,绘制标准曲线。结果表明:硝基呫吨酮质量浓度为0.005~1 mg/L时,线性回归方程为y=322 219x+ 1 454.3,相关系数R2=0.999 6。符合r>0.99的要求[6]。标准曲线见图3。
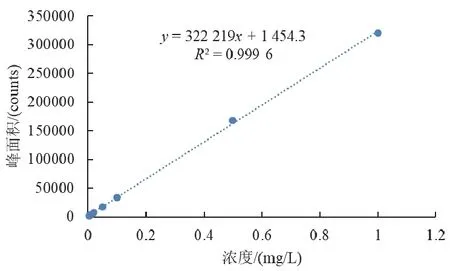
图3 硝基呫吨酮标准曲线
2.4 准确度试验
称取约0.5 g不含硝基呫吨酮的硝磺草酮原药(样品A),加入适量乙腈溶解,加入一定量的硝基呫吨酮标准品,使其添加浓度分别为0.1、2、5 mg/kg,稀释至5 mL,每个浓度配置2个平行样品。在上述1.3色谱条件下测定其含量,计算硝基呫吨酮的回收率平均值为86%~100%。根据准则要求,当杂质含量<0.1%时,回收率应为75%~125%[6]。根据测试结果,添加回收率满足准则要求。回收率数据见表2。
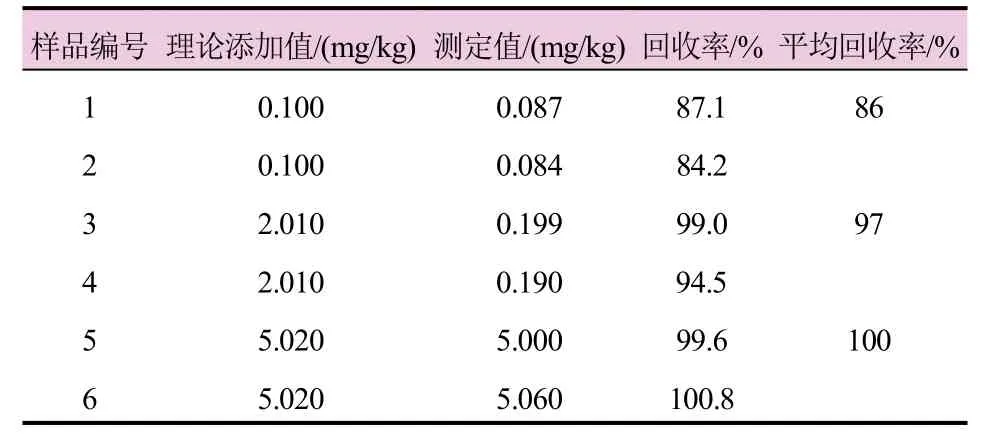
表2 硝基呫吨酮回收率测定结果
2.5 精密度试验
对硝磺草酮样品B中的硝基呫吨酮进行6次平行测定,测得硝基呫吨酮含量的标准偏差为0.022,相对标准偏差为3.30%。根据霍维茨方程相对偏差应小于11.36% (%RSDr=2(1-0.5log0.00000068)×0.67=11.36)[6]。由测定结果可知,精密度满足准则要求。精密度数据见表3。
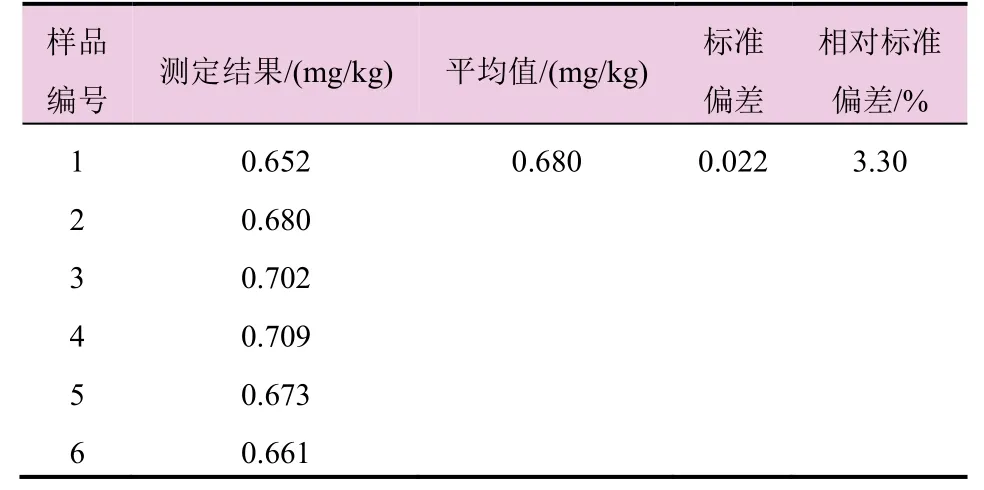
表3 硝基呫吨酮精密度测定结果
2.6 最低定量限(LOQ)
10 μg/L的硝基呫吨酮的标样的信噪比为11,大于10倍信噪比[6]。其浓度折合到硝磺草中的质量分数为0.1 mg/kg,且该浓度的回收率符合要求。因此0.1 mg/kg浓度适合作为该方法的LOQ。
3 结 论
本方法在综合文献的基础上对国标GB 29382—2012中的硝基呫吨酮的分析方法进行了改进,包括采用的离子源类型、扫描的极性、扫描的离子对、流动相中乙酸铵的浓度、流速以及将单点外标定量改为标准曲线外标定量。
改进后的方法扩大了定量范围,缩短了分析时间,提高了分析效率。试验数据显示,该方法专属性良好,灵敏度、准确度和精密度高,线性范围广,适合用于硝磺草酮中硝基呫吨酮的定量分析。该方法可为生产企业检测该杂质提供方法参考。
猜你喜欢
中国化工贸易·下旬刊(2019年10期)2019-10-21
农家科技(2019年1期)2019-03-13
科学家(2016年17期)2017-10-17
食品界(2017年7期)2017-08-24
价值工程(2016年29期)2016-11-14
科技视界(2016年22期)2016-10-18
当代化工(2016年3期)2016-07-10
中国信息化·学术版(2013年3期)2013-06-25
