
白细胞介素33通过增强炎症反应加重脂多糖诱导的急性肾损伤*
2022-09-01 10:07董一飞
中国病理生理杂志 2022年8期
付 阳,董一飞
(南昌大学第二附属医院心血管内科,江西南昌 330006)
急性肾损伤(acute kidney injury,AKI)是一种常见危重症,是指肾功能在短时间内突然下降引起血清肌酐(serum creatinine,SCr)和血尿素氮(blood urea nitrogen,BUN)显著升高的临床综合征,可由多种病因引起,如脓毒症、休克和心肌梗死等[1-3]。AKI的发病率和死亡率在世界范围内呈逐渐升高趋势,每年大约有近200 万的病死率,造成严重的医疗安全问题[4]。一般来说,AKI 的病程是短程的,但长时间患病会引起肾脏功能障碍,并最终发展成为慢性肾病(chronic kidney disease,CKD)[5]。脓毒症(sepsis)是引起AKI 的主要原因之一,会导致微血管功能障碍、炎症和小管损伤等许多病理改变[6-7]。目前对AKI 的病理机制研究已很透彻,但临床治疗手段有限,因此,亟需寻找新的、有效的AKI治疗方法。
白细胞介素33(interleukin-33,IL-33)是新近发现的一种细胞因子,属于IL-1 家族,可由上皮细胞、巨噬细胞、平滑肌细胞、树突状细胞、间质成纤维细胞等多种细胞合成分泌[8-12]。当IL-33 与其受体ST2结合后,能发挥多种生物学特性[13]。有报道指出,IL-33通过增强炎症反应和增加微血管通透性加重脂多糖(lipopolysaccharide,LPS)诱导的小鼠急性肺损伤[14]。而另有其它研究显示,IL-33 在一些炎性疾病中具有保护作用[15]。目前,对IL-33 在炎性疾病中的确切作用机制仍不清晰,有待进一步研究。
本研究拟采用LPS 构建脓毒症所致AKI 小鼠模型,探索IL-33在AKI中的作用及其可能的机制。
材料和方法
1 动物和试剂
6~8周龄雄性C57BL/6小鼠,体重(22±3)g,购自湖南斯莱克景达实验动物有限公司;IL-33 购自Enzo Life Sciences;LPS购自Sigma;IL-1β、IL-6和肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)ELISA 试剂盒购自Invitrogen;蛋白提取试剂盒购自苏州碧云天生物技术研究中心;抗IL-1β、IL-6、TNF-α、核苷酸结合寡聚化结构域样受体蛋白3(nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)、IL-33 和核因子κB(nuclear factor-κB,NF-κB)抗体均购自Abcam;相应Ⅱ抗购自Proteintech。
2 动物模型
小鼠饲养在南昌大学江西医学院动物管理中心,光照/黑暗周期为12 小时,饮水和食物自由。饲养环境温度保持在(22±3)℃,湿度保持在(54±6)%。小鼠适应性饲养环境约7 d。然后将小鼠随机分为3组:(1)control组(n=8):小鼠在整个实验过程中均给予腹腔注射磷酸盐缓冲液(phosphate-buffered saline,PBS),不使用其他试剂;(2)LPS 组(n=8):小鼠腹腔注射等体积的PBS,6 h 后再注射LPS(10 mg/kg),24 h 后处死小鼠;(3)LPS+IL-33 组(n=8):小鼠先腹腔注射IL-33(50 µg/kg),6 h 后再注射LPS(10 mg/kg),24 h 后处死小鼠。注射试剂的方式和剂量参照以前的研究[16-17]。小鼠处理流程总结见图1。所有小鼠的处理均经南昌大学第二附属医院动物管理委员会批准,实验均按照相关指南和规定进行。

Figure 1.A summary of treatment procedure in mice.PBS:phosphate-buffered saline;LPS:lipopolysaccharide;IL-33:interleukin-33.图1 小鼠处理流程总结示意图
3 SCr和BUN测定
处死小鼠后,用真空管从右心室取得全血。然后用离心机在4 ℃、4 000×g离心30 min,提取出血清。采用BS-800化学分析仪(迈瑞公司)自动检测各组肾损伤标志物SCr和BUN水平。
4 ELISA法检测血清中炎症因子水平
血清中炎症因子IL-1β、IL-6 和TNF-α 水平的检测采用相应的ELISA 试剂盒。具体操作流程按试剂盒提供的实验说明进行。
5 HE染色
收集的肾组织立即用4%甲醛固定,经酒精脱水后,采用石蜡包埋(5µm),然后用苏木精/伊红染色,最后用光学显微镜观察组织切片。肾损伤评分用于评估肾损伤程度。
6 Western blot法检测相应蛋白表达变化
采用蛋白提取试剂盒提取肾组织的总蛋白溶液,按比例将总蛋白溶液和上样缓冲液均匀混合,置于100 ℃水中10 分钟使其变性,蛋白上样量为50µg。使用10%的SDS-PAGE 分离总蛋白,然后将分离的蛋白通过转膜至PVDF 膜上,最后用5%的牛奶封闭该膜1 h。特定的蛋白条带采用相应的一抗在4 ℃冰箱中孵育约24 h,取出后用相应的二抗在室温条件下孵育约1 h。最后,使用增强型化学发光检测试剂盒和相应的扫描仪来检测蛋白质条带。
7 统计学分析
采用GraphPad Prism 7.0 软件进行统计分析。实验数据均以均数±标准差(mean±SD)表示。多组间均数比较采用单因素方差分析。以P<0.05 为差异有统计学意义。
结果
1 LPS能增加肾组织中IL-33的表达
如图2 所示,小鼠肾组织IL-33 蛋白表达水平在LPS 刺激后显著增加。该结果证明,IL-33 的表达与LPS的刺激密切相关。
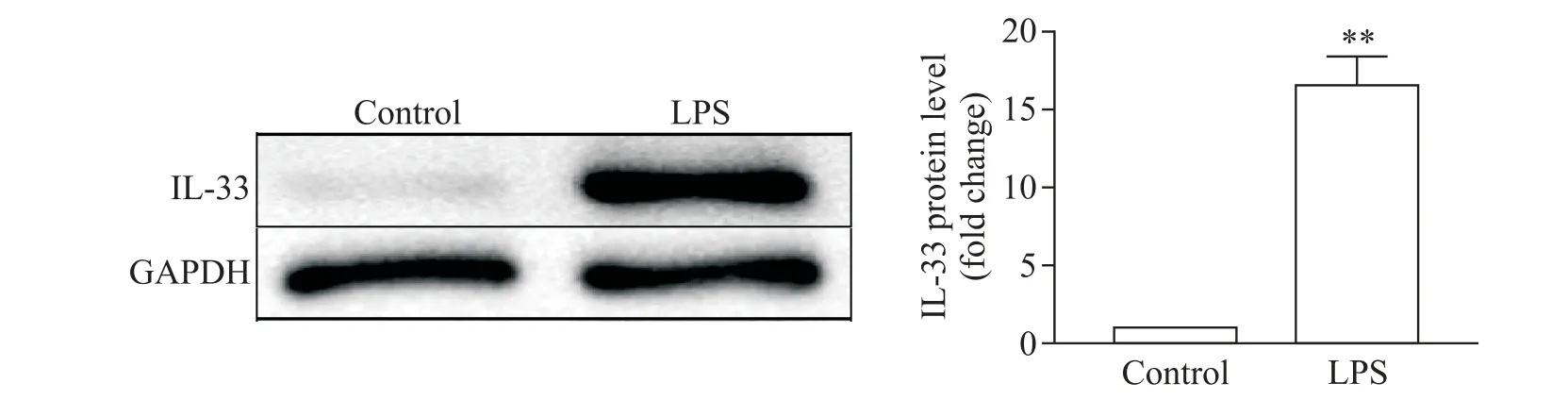
Figure 2.The protein expression of interleukin-33(IL-33)was markedly up-regulated by stimulation of lipopolysaccharide(LPS).The protein expression of IL-33 was detected by Western blot.Mean±SD. n=8.**P<0.01 vs control group.图2 LPS能增加小鼠肾组织中IL-33的表达
2 IL-33预处理显著加重LPS诱导的AKI
如图3A、B所示,与control组相比,LPS组中小鼠BUN 和SCr 水平在LPS 刺激后显著升高;而给予IL-33 预处理后,LPS+IL-33 组小鼠BUN 和SCr 水平进一步升高,表明IL-33 预处理可明显加重LPS 刺激引起的AKI。同时,ELISA 检测结果如图3C~E 所示,血清中炎症因子IL-1β、IL-6 和TNF-α 水平变化与BUN 和SCr 的变化一致,说明IL-33 预处理能明显加重LPS刺激引起的全身炎症反应。上述结果提示,IL-33 预处理可显著加重LPS诱导的AKI。
3 IL-33预处理加重LPS所致小鼠肾组织病理损伤
如图4 所示,与control 组相比,LPS 组小鼠肾组织结构明显被破坏,肾组织水肿明显,而IL-33 预处理显著增强了LPS 的肾损伤作用。该结果提示,IL-33 预处理能显著加重LPS 刺激所致的肾组织病理损伤。
4 IL-33预处理通过增强炎症反应加重LPS诱导的AKI
如图5 所示,与control 组相比,LPS 组小鼠肾组织中炎症因子(IL-1β、IL-6、TNF-α 和NLRP3)表达水平显著升高,而给予IL-33 预处理后,小鼠肾组织中炎症因子(IL-1β、IL-6、TNF-α 和NLRP3)表达水平进一步升高。上述结果表明IL-33 预处理可通过增强炎症反应进一步加重LPS诱导的AKI。
5 IL-33预处理通过进一步激活NF-κB信号通路增强炎症反应
如图6 所示,与control 组相比,LPS 组小鼠肾组织NF-κB p65 磷酸化水平显著升高,而IL-33 预处理后NF-κB p65 磷酸化水平进一步升高。这一结果表明,IL-33 预处理可能是通过进一步激活NF-κB 信号通路增强炎症反应。
讨 论
AKI 是一种以肾功能损害为特征的复杂性疾病。根据之前的研究显示,急性肾功能衰竭(acute kidney failure,AKF)的主要病因是脓毒症,而脓毒症与AKI 的结合使AKF 的死亡率从45%显著提高到70%[1,18]。此外,越来越多的证据表明AKI 患者在长期可能发展成为CKD[5]。然而,迄今为止尚无有效的AKI 治疗策略,所有亟需我们进一步探索脓毒性AKI的发病机制,并研究新的预防和治疗措施。

Figure 3.Pretreatment with interleukin-33(IL-33)aggravated the renal dysfunction and systemic inflammation of acute kidney injury mice induced by lipopolysaccharide(LPS).A and B:the levels of renal injury biomarkers,blood urea nitrogen(BUN)and serum creatinine(SCr);C,D and E:the serum levels of inflammatory cytokines,IL-1β,IL-6 and tumor nwcrosis factor-α(TNF-α),were assessed by ELISA kits.Mean±SD. n=8.*P<0.05 vs control group;#P<0.05 vs LPS group.图3 IL-33预处理加重LPS诱导的AKI小鼠肾功能不全和全身炎症反应
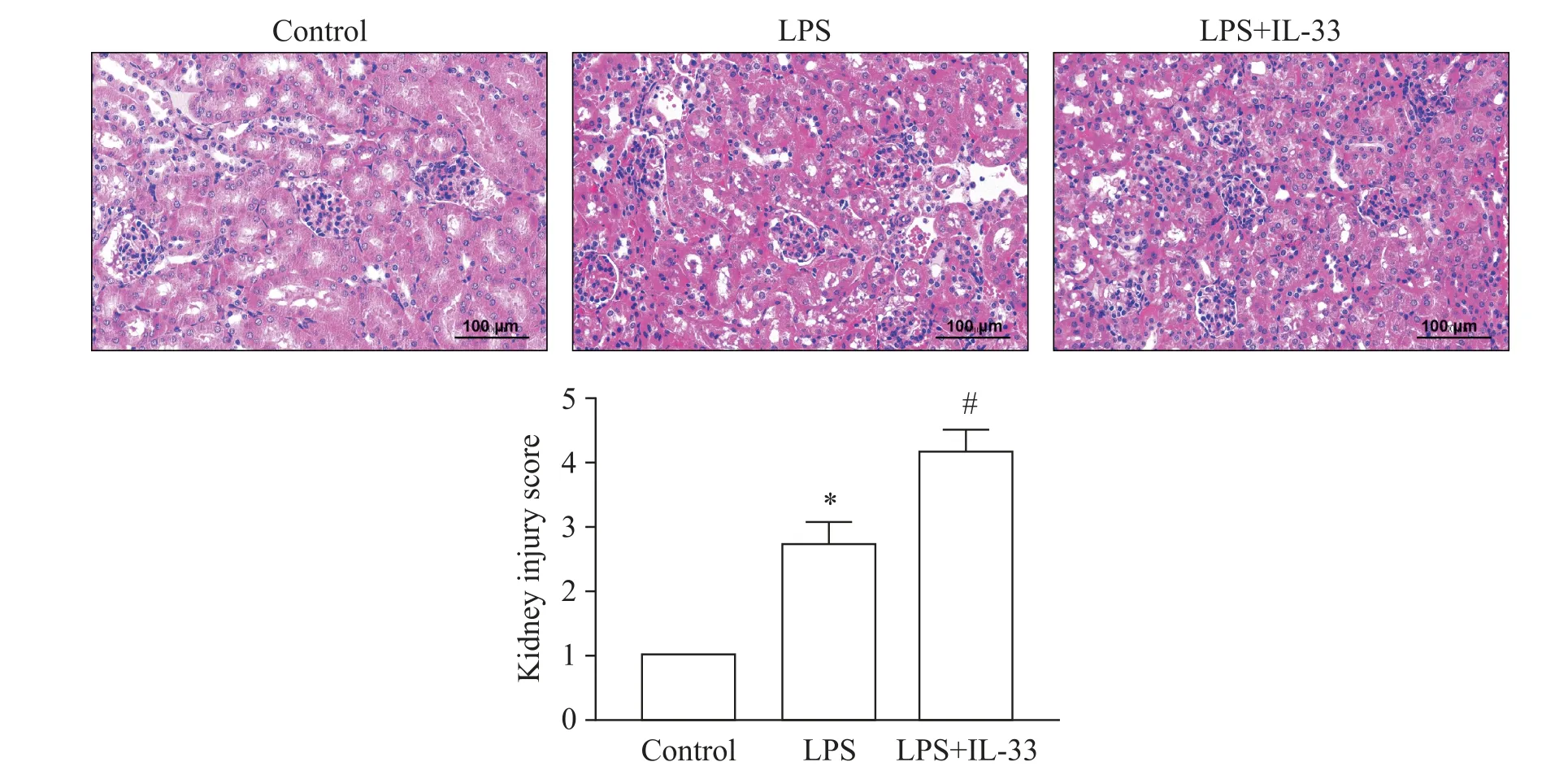
Figure 4.Pretreatment with interleukin-33(IL-33)aggravated the histopathological injury of kidney tissues induced by lipopolysaccharide(LPS).Mean±SD. n=8.*P<0.05 vs control group;#P<0.05 vs LPS group.图4 IL-33预处理加重LPS所致肾组织病理损伤
IL-33,也被称为IL-1F11 和NF-HEV,于2005 年首次被确定为IL-1 家族的新成员[19-20]。与传统的细胞因子不同,IL-33 对炎症反应具有双重作用,这主要是由于IL-33 的不同存在形式(如细胞内核因子或细胞因子)[21]。当IL-33 作为细胞内核因子时,它可以隔离转录因子NF-κB,从而抑制NF-κB 表达,最终减轻炎症反应[22]。而作为细胞因子时,IL-33 可通过与ST2 受体结合来调节免疫应答。IL-33 和ST2 受体结合对不同T 细胞的分化和功能产生显著影响,尤其是调节2 型免疫应答。例如,IL-33 可以增加Th2免疫细胞的数量,并促进Th2 细胞的细胞因子或趋化因子的过度表达,如IL-13、IL-9 和IL-5[23]。IL-33还可以促进Th1 细胞和CD8+细胞毒性T 淋巴细胞的扩增,表现出抗病毒作用[24-25]。另据报道,IL-33 可通过增强炎症反应从而加重LPS 诱导的小鼠急性肺损伤[14]。而其他一些研究表明,IL-33 在某些炎症性疾病中具有保护作用[15]。因此,IL-33 的确切作用机制仍不明确。在本研究中,我们探讨了IL-33 对LPS 诱导的小鼠AKI的影响。

Figure 5.Pretreatment with interleukin-33(IL-33)enhanced renal inflammation induced by lipopolysaccharide(LPS).The protein expression of IL-1β,IL-6,tumor nwcrosis factor-α(TNF-α)and nucleotide-binding oligomerization domain-like receptor protein 3(NLRP3)in kidney tissues was detected by Western blot.Mean±SD. n=8.*P<0.05 vs control group;#P<0.05 vs LPS group.图5 IL-33预处理能增强炎症反应,进一步加重LPS诱导的AKI
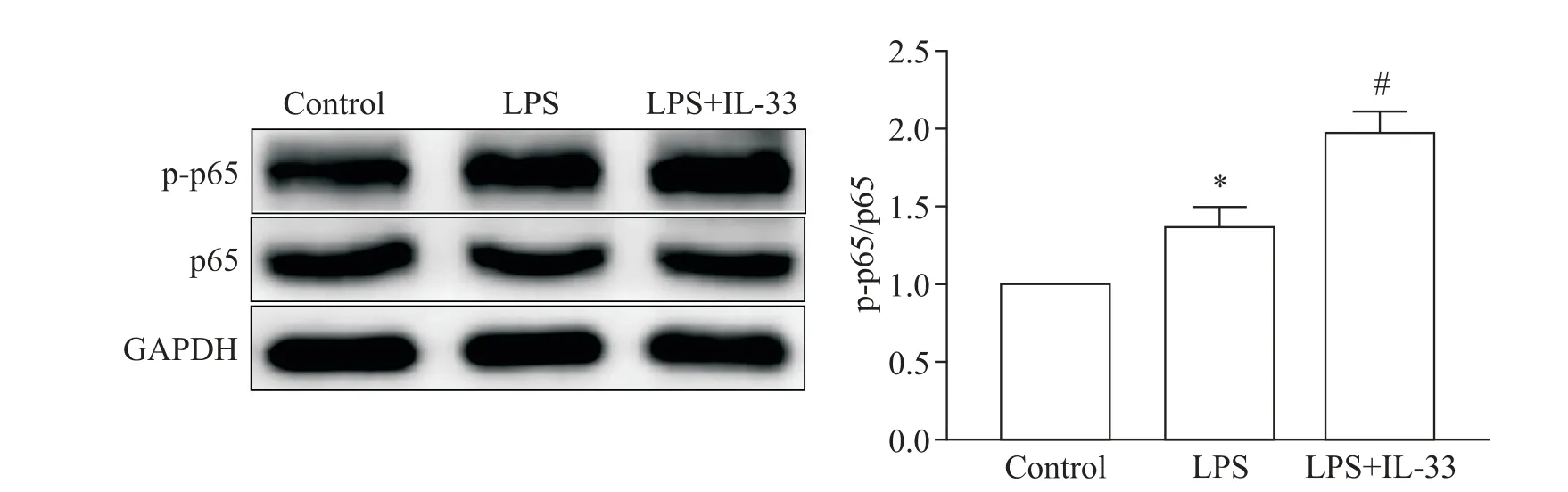
Figure 6.Pretreatment with interleukin-33(IL-33)enhanced inflammation by expediting the activation of nuclear factor-κB(NF-κB)signaling pathway.Mean±SD. n=8.*P<0.05 vs control group;#P<0.05 vs lipopolysaccharide(LPS)group.图6 IL-33预处理通过进一步激活NF-κB信号通路增强炎症反应
在本研究中,我们探讨了IL-33 对LPS 刺激引起AKI 的影响,并进一步深入研究了其可能存在的分子机制。多项研究表明,IL-33 的表达与LPS 的刺激密切相关,我们的研究再次证实了IL-33 的表达与LPS 的刺激密切相关。我们构建了LPS 刺激诱导的AKI模型小鼠,以IL-33预处理后,血清BUN和SCr水平进一步升高,表明IL-33 能使LPS 所致AKI 的肾功能进一步受损。同时,IL-33 预处理后,血清中IL-1β、IL-6 和TNF-α 等炎症因子水平也进一步增加,表明IL-33 能明显加重LPS 刺激引起全身炎症反应。HE染色结果显示,LPS刺激破坏了肾脏结构,肾组织出现水肿,而IL-33 预处理进一步增强了这种作用。我们还发现,IL-33 预处理显著加重了AKI 的炎症水平,表现为炎症因子如IL-1β、IL-6、TNF-α 和NLRP3的蛋白表达水平比单纯LPS 刺激增加更为显著。最后,我们研究了IL-33 在NF-κB 信号通路中的作用,结果表明,IL-33 预处理可进一步激活NF-κB 信号通路。所有这些数据表明,IL-33 可通过增强炎症反应加重LPS诱导的AKI,其分子机制可能是进一步激活了肾脏NF-kB信号通路。
综上所述,本研究发现IL-33 对LPS 诱导的AKI具有加重损伤的作用。进一步研究提示IL-33 能通过促进炎症反应加重LPS诱导的AKI,其分子机制可能是进一步促进了肾脏NF-kB 信号通路的激活。本研究提示IL-33 可能成为脓毒症所致AKI 或者其他肾脏炎性疾病的新干预靶点。
猜你喜欢
中国中医急症(2022年9期)2022-10-12
材料与冶金学报(2022年2期)2022-08-10
温州大学学报(自然科学版)(2022年2期)2022-05-30
现代临床医学(2022年2期)2022-04-19
湖南畜牧兽医(2021年6期)2022-01-24
食品安全导刊(2021年21期)2021-08-30
猪业科学(2021年5期)2021-06-02
中国畜禽种业(2021年4期)2021-05-21
滨州医学院学报(2021年2期)2021-05-13
建材发展导向(2021年23期)2021-03-08
