
First-principles study of a new BP2 two-dimensional material
2022-08-31 09:55:28ZhizhengGu顾志政ShuangYu于爽ZhirongXu徐知荣QiWang王琪TianxiangDuan段天祥XinxinWang王鑫鑫ShijieLiu刘世杰HuiWang王辉andHuiDu杜慧
Chinese Physics B 2022年8期
Zhizheng Gu(顾志政) Shuang Yu(于爽) Zhirong Xu(徐知荣) Qi Wang(王琪)Tianxiang Duan(段天祥) Xinxin Wang(王鑫鑫) Shijie Liu(刘世杰)Hui Wang(王辉) and Hui Du(杜慧)
1Henan Key Laboratory of Photoelectric Energy Storage Materials and Applications,School of Physics and Engineering,Henan University of Science and Technology,Luoyang 471023,China
2State Key Laboratory of Superhard Materials,Jilin University,Changchun 130012,China
Keywords: two-dimensional material,density functional theory,direct band gap,strain
1. Introduction
Since graphene was synthesized in 2004,[1]twodimensional(2D)materials have received extensive attention.In recent years, researchers have made a series of important progress in 2D materials, and discovered many new 2D materials. The common 2D materials can generally be classified as follows: transition metal dichalcogenides,[2,3]transition metal oxides,[4,5]and graphene analogues,[6,7]2D V–V binary materials.[8]Two-dimensional materials have diverse band structures, covering various types of insulators,[9,10]semiconductors,[11–13]semimetal,[1,14,15]conductors,[16]and superconductors.[17]In the fields of electronic components,[18]catalysts[19]and photovoltaic solar energy,[20]2D materials have already displayed practical applications.
Bulk boron phosphide (BP) is a typical semiconductor material with wide band gap, which has a wide range of applications in solid-state neutron detectors and other fields.[21]Recently, researchers have synthesized large-size sphalerite BP single crystal,[22]which not only has very high hardness,good thermal stability,high thermal conductivity,but also has a suitable band gap (about 2 eV). Therefore, BP is considered to be an ideal material for realizing a new generation of semiconductor devices under extreme conditions.[23–25]The 2D material of B–P system has also attracted people’s attention. Theoretically, Wanget al.[26]predicted a hexagonal 2D BP structure. Liuet al.[27]and Wanget al.[28]reported several novel 2D materials in BP5, which have excellent electronic properties and good mechanical properties. Experimentally,a 2D hexagonal BP film was fabricated on SiO2and AlN substrates.[29]These results have made us very curious about the 2D B–P system,and we are eager to explore whether it has other new materials with better performance.
Here, we have conducted a systematic study on the 2D BP system and predicted a new 2D material(PMM2)by using first principles.The phonon spectrum and molecular dynamics(MD) simulation results show that the structure has dynamic stability and good thermal stability(above 600 K).The PMM2 structure has an indirect band gap. When a suitable strain is applied, the structure can exhibit other electronic properties,such as direct band gap semiconductor and metal. The above excellent performance also makes the PMM2 have potential application prospects in optoelectronic devices.
2. Methods
For the 2D structure search of BP2system, we utilize the CALYPSO code.[30,31]We use Viennaab initiosimulation package (VASP)[32,33]for structural optimization, electronic properties and other related calculations. We use the projector augmented wave (PAW) method[34]to treat the ion potential (2s2p1for B and 3s23p3for P). In order to ensure the accuracy of the calculation, we use the cutoff energy of 600 in all calculations, and we integrate the reciprocal space with a resolution of 2π×0.03 ˚A−1by using the Monkhorst–Pack Brillouion zone scheme.We use the Conjugated gradient algorithm to relax the 2D PMM2 structure with the force components of 10−3eV·˚A−1,and the convergence criterion of the total energy is 1.0×10−6eV.In order to avoid the influence of adjacent layers,we add a 15 ˚A vacuum layer to the 2D structure. We use phonopy code to calculate the phonon spectrum of the 2D structure to determine its dynamic stability.[35]Firstprinciples MD simulations are conducted with the canonical ensemble(NVT)ensemble at different temperatures to further study the thermal stability of the 2D PMM2 structure,including 300 K, 400 K, 500 K, 600 K, 700 K, 800 K and 900 K.In order to ensure the accuracy of the calculation,a 4×6×1 supercell (144 atoms) are used. The first-principle molecular dynamics simulations lasted for 8 ps with the time step of 1 fs.
3. Results and discussion
Using the CALYPSO software, we carry out a detailed structure search and design of the BP2system, and obtain a new 2D layered structure (space group PMM2), as shown in Fig. 1. The lattice constants of the PMM2 structure area=3.2552 ˚A andb=4.9839 ˚A, respectively. The structure is not a single atomic layer structure,but has a certain spatial configuration with a thickness of 3.2119 ˚A. All the B atoms are equivalent and form a stable four-coordinate configuration.On the other hand,there are two kinds of equivalent atoms for the P atom, which form three-coordinate and four-coordinate in space,respectively. In this layered structure,there are three types of bonding, including B–B, P–P, and B–P, and the corresponding bond lengths are 1.797 ˚A, 2.174 ˚A, and 2.017 ˚A,respectively.

Fig.1. Top(a)and side[(b),(c)]views of the 2D PMM2. The big and small spheres are P and B atoms,respectively.
Next,we calculate the phonon spectrum of the 2D PMM2 structure to verify its dynamic stability. As shown in Fig. 2,it can be observed that there is no imaginary frequency in the entire Brillouin zone, which also indicates that the 2D PMM2 structure is dynamic stable. In order to further evaluate the mechanical stability of the 2D PMM2 structure, we calculate the linear elastic constants asC11=83.8 GPa,C22=124.9 GPa,C12=22.2 GPa andC66=42.6 GPa. According to the Born–Huang standard,[36]they meet the conditions ofC11C22−C212>0 andC66>0, which indicate that the 2D PMM2 structure is mechanical stable.

Fig.2. Phonon spectra(left)and partial phonon density of state(right)of PMM2 sheets.
For an ideal material, excellent thermal stability is also an essential requirement. First-principles MD simulations are conducted with the canonical ensemble (NVT) ensemble at different temperatures to further study the thermal stability of the 2D PMM2 structure. As shown in Fig.3(a),at 300 K,the bond length and bond angle of the structure change slightly,but they are not enough to destroy the original chemical bond.In order to further study the temperature effect on the stability of the structure, we conduct MD studies at higher temperatures. As shown in Fig. 3(b), when the temperature rises to 600 K, although the position of each atom fluctuates greatly,the structure can maintain the original bond style. When the temperature is 700 K,the B–B bond begins to break.

Fig.3. The MD simulations for the PMM2 at the temperature of 300 K(a) and 600 K (b). The insets are the top and side views of the 2D PMM2 structure at the end of the MD simulation.
Ideal nanomaterials generally need to have good mechanical properties. The evolutions of the stress versus the strains are calculated to characterize mechanical properties of the 2D PMM2. The strain is defined by the following formula:(m −m0)/m. Themis the lattice parameter when there is strain, and them0is the lattice parameter when there is no strain. First,we calculate the stress versus strain curve of the 2D PMM2 under biaxial strain,as shown by the blue curve in Fig.4.The compression and tension biaxial strains that the 2D PMM2 can withstand are−4% and 14%, respectively. Subsequently, we calculate the data of the PMM2 under uniaxial strain.Since the two axial directions in the plane are not equivalent,the stress–strain curves are calculated along theaandbdirections, as shown in Fig. 4. In the specific calculation of uniaxial strain,we apply strain in one of the directions,while the lattice in the other direction is allowed to relax freely. The maximum compressive strain the 2D PMM2 can withstand is 8%whether it is in theaorbdirection.When a tensile strain is added,the maximum tensile strain in thea-axis is 12%,and the corresponding maximum stress is 2.85 GPa, while the maximum tensile strain in theb-axis is 24%,and the corresponding maximum stress is 5.12 GPa. The above data shows that the mechanical properties of the 2D PMM2 structure are very excellent,similar to graphene and MoS2.[37,38]
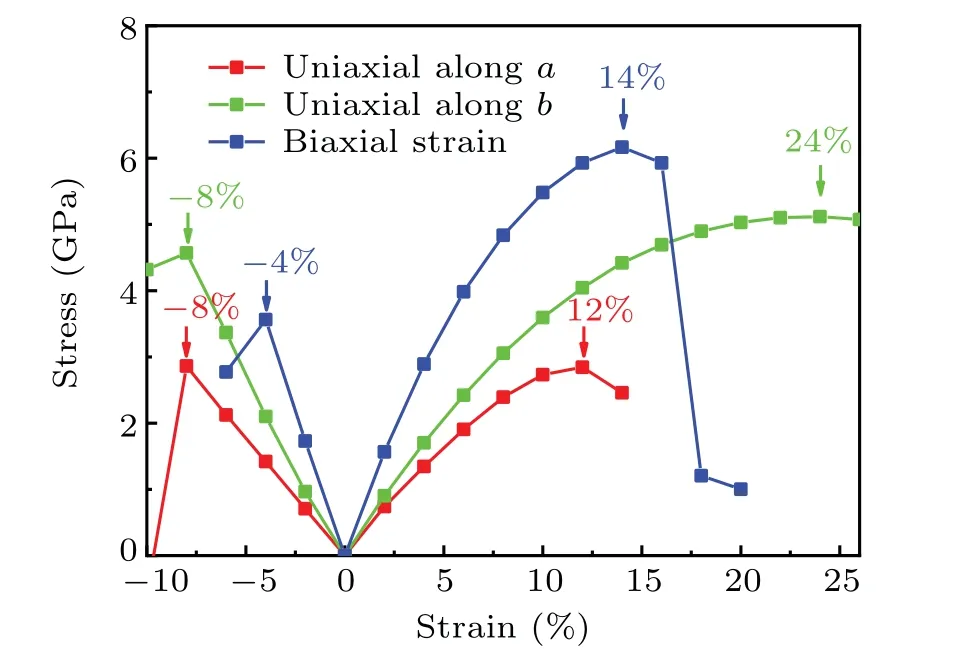
Fig.4. The evolution of the stress in the 2D PMM2 versus the biaxial and uniaxial strains.
To characterize the electronic properties of the PMM2 structure,we calculate the band structures and density of states(DOS).The band structure of the 2D PMM2 is obtained by the PBE method, as shown in Fig. 5 (the left panel black curve),which has an indirect band gap of 0.11 eV. Next, we use the HSE06 method[39]to recalculate the band structure of PMM2 in order to obtain a more accurate value of the band gap, as shown in the red curve on the left of Fig. 5. The calculation results show that the PMM2 has a band gap with 0.79 eV,and band structures obtained by the two methods have similar shapes. Subsequently,we calculate the DOS of the 2D PMM2 structure. The PDOS demonstrates that the VBM and CBM are mainly contributed by p-orbital of phosphorus and boron atoms. As shown in Fig. S3, the calculated ELF shows that there is a covalent interaction between P and P,B and P.

Fig.5.The calculated band structures(left panel)of 2D PMM2 at PBE level(black),HSE level(red)and the DOS(right panel)using PBE method.
Uniaxial or biaxial strain can effectively adjust the electronic properties of the structure, and this method has been widely confirmed in experiments.[40,41]According to the stress–strain curve in Fig.4,we have obtained the stable strain range of the 2D PMM2 structure. Next,we carry out the study of strain-tuning the band gap of PMM2.

Fig.6. Band gap vs in-plane biaxial strain(a)and uniaxial strain along the a direction(b)for the PMM2.
First, we conduct a study on the electronic properties of 2D PMM2 under different biaxial strains. Figure 6 shows that when the tensile strain is applied,the PMM2 structure still has an indirect band gap; while the band gap of the 2D PMM2 structure gradually increases and reaches the maximum when the strain reaches 8%, and then starts to decrease approximately linearly. When the strains are 8%–14%, the PMM2 exhibits a direct band gap of 1.39–0.93 eV at HSE06 (0.57–0.15 eV at PBE).Under the control of compressive strain,the bandgap of PMM2 gradually decreases. Uniaxial strain is easier than biaxial strain to change the symmetry of the material and to obtain new properties. First, we calculate the regulation effect of uniaxial strain along thea-axis on the electronic properties. Figure 6(b)shows that the regulation of electronic properties by uniaxial strain is very similar to that of biaxial strain. When the strains are 8%–12%, the PMM2 exhibits a direct band gap of 1.14–0.85 eV at HSE06 (0.38–0.10 eV at PBE).Under the control of compressive strain,the bandgap of PMM2 gradually decreases. We also study strain effect alongb-axis as shown in Fig.S1 in the supplementary materials. In the entire strain range of−8%to 24%,the structure has always maintained an indirect band gap.
Next, we compare the band structure under different biaxial strains to obtain the physical mechanism of strain regulating electronic properties, as shown in Fig. 7. When no strain is applied,PMM2 has an indirect band gap. We denote the conduction band minimum(CBM)and valence band maximum(VBM)of the PMM2 by points A and C,and the second high valence band and second low conduction band by B and D, respectively. When the biaxial strain gradually increases,point B begins to gradually increase, and point D begins to decrease. When the biaxial strain reaches 8%, points B and D become the new CBM and VBM,which also indicates that the PMM2 has been transformed into a direct band gap semiconductor. When a larger strain is applied to the PMM2, the energy at point D gradually decreases, so there is a tendency for the band gap to decrease. The regulation mechanism of uniaxial strain on electronic properties is very similar to that of biaxial strain(see Fig.S2 in the supplementary materials).
Two-dimension ferroelectricity has attracted intensive interest over the past several years.[42]By carefully analyzed the structural features of the PMM2 structure, we find that the monolayer is non-centrosymmetric, and the electronegativity of phosphorus atoms is greater than that of boron atoms. As a result,a spontaneous polarization occurs in PMM2 monolayer with the direction of polarization along thebdirection.The estimated intensity of polarization is 0.74×10−12C/m by using the Berry phase approaches,[43,44]which also indicates that the BP2structure is a two-dimensional ferroelectric material.
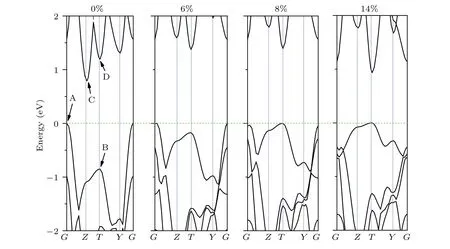
Fig.7. Biaxial strain manipulated indirect-to-direct band gap of 2D PMM2.
4. Conclusion and perspectives
In summary, we have predicted a new 2D PMM2 structure, which has an indirect band gap. The analysis of the phonon dispersive curves shows that the 2D PMM2 is dynamic stable. The study of molecular dynamics shows that the 2D PMM2 can be stable under high temperature, even at 600 K.Most importantly,when a suitable strain is applied,the structure can exhibit other electronic properties,such as direct band gap semiconductor. In addition, the small strain can tune the band gap value of the PMM2 structure to around 1.4 eV,which is very close to the ideal band gap of solar materials. The electronic properties of the adjustable band gap and excellent mechanical properties make the 2D PMM2 structure have a good application prospect in the field of optoelectronic devices and photovoltaic materials.
Acknowledgements
Project supported by the National Natural Science Foundation of China (Grant Nos. 12004102 and 11847094),the China Postdoctoral Science Foundation(Grant No.2020M670836),the Open Project of State Key Laboratory of Superhard Materials in Jilin University(Grant No.201703),and Student Research Training Program of Henan University of Science and Technology(Grant No.WLSRTP202118).
猜你喜欢
辽河(2022年12期)2023-01-29 13:24:58
Chinese Physics B(2022年12期)2022-12-28 09:55:32
文萃报·周二版(2022年28期)2022-07-14 09:49:40
文萃报·周二版(2022年25期)2022-06-23 10:29:09
火花(2021年10期)2021-11-04 09:23:52
Chinese Physics B(2021年10期)2021-10-28 07:12:22
下一代(2021年2期)2021-07-05 02:46:10
东坡赤壁诗词(2020年3期)2020-07-04 02:50:05
理论与创新(2019年5期)2019-09-10 07:22:44
领导文萃(2018年24期)2018-01-15 07:57:36

- Chinese Physics B的其它文章
- Magnetic properties of oxides and silicon single crystals
- Non-universal Fermi polaron in quasi two-dimensional quantum gases
- Purification in entanglement distribution with deep quantum neural network
- New insight into the mechanism of DNA polymerase I revealed by single-molecule FRET studies of Klenow fragment
- A 4×4 metal-semiconductor-metal rectangular deep-ultraviolet detector array of Ga2O3 photoconductor with high photo response
- Wake-up effect in Hf0.4Zr0.6O2 ferroelectric thin-film capacitors under a cycling electric field