
Matrin-3 dysfunction in myopathy and motor neuron degeneration
2022-08-15 15:46:18CarolineWardUdaiBhanPandey
中国神经再生研究(英文版) 2022年3期
Caroline Ward, Udai Bhan Pandey
The MATR3 protein was first identified comprising the nuclear matrix, an essential part of preserving the skeletal framework of the nucleus, amongst other nuclear matrins.Composed mainly of intrinsically disordered regions, the protein is made up of 847 amino acids, creating 4 distinct functional domains:two zinc-finger domains (ZF1 and ZF2) and two RNA-recognition motifs (RRM1 and RRM2,) as well as a highly acidic carboxy-terminal with histone-binding ability.This structural design allows MATR3 protein to interact with DNA and/or DNA/RNA-binding proteins, aiding in chromosomal and genomic integrity regulation,RNA-binding mediated post-transcriptional mRNA regulation, and nuclear lamina association to maintain nuclear framework.An autosomal dominant Ser85Cys (S85C) mutation in MATR3 was identified in a multigenerational family displaying vocal cord and pharyngeal weakness as well as distal and asymmetrical myopathy (Senderek et al., 2009).Intriguingly,a neurogenic or myopathic pattern was indicated by these patients neurophysiological and muscle biopsy-based examinations.They displayed several signs emblematic of upper motor neuron lesions such as a “split-hand”pattern, tongue fasciculations, brunt jawjerk, upper limb reflexes, and brisk knee.Due to this, S85C patients in three independent families were reassessed and found to suffer progressive respiratory failure leading to death 15 years after onset (Johnson et al.,2014), in contrast to the typical 2–5 years.This led to a recategorization of S85C-associated disorder to “slow progressive amytrophic lateral sclerosis (ALS) with distal myopathy,”and is approximated at 0.5% frequency of all mutations.Initially, 3 cohorts containing unique missense MATR3 mutations associated with ALS were characterized; Phe115Cys (F115C)and Thr622Ala (T622A) were found in familial ALS patients, and Pro154Ser (P154S) in a sporadic ALS patient (Johnson et al., 2014).Subsequently, 3 additional MATR3 mutations including p.Ser610Phe (Xu et al., 2016), a missense mutation found in ALS patients from Chinese origin with 3 variants (p.Ala313Gly,p.Arg147Lys, and p.Gln347Lys).Secondly, a duplication of exon 15 referred to as MATR3 Variant 5 (Castro et al., 2020) was found in a patient presenting symptoms of frontotemporal dementia, lastly, a heterozygous missense mutation called Ala72Thr was found in a Taiwanese ALS patient (Lin et al., 2015).
Recently, multiple cellular and animal models of MATR3 have been generated to investigate the molecular basis of disease-causing mutations associated with distal myopathy and ALS.Expression of MATR3 fusion protein(MATR3-YFP) in human neuroglioma H4 cells and Chinese hamster ovary epithelial cells demonstrated through immunostaining and imaged fluorescently tagged proteins, that wildtype and mutant MATR3 proteins are restricted mainly to the nucleus in a punctate pattern even under stress (Gallego-Iradi et al.,2015).While similar in structure and function to other RNA-binding proteins connected to ALS such as FUS and TDP-43 these proteins are largely recruited to stress granules under stressful conditions, MATR3 however, reacts differently.In an analysis of patient-derived muscle biopsy samples and fibroblasts containing S85C mutation, MATR3 protein was not found to be incorporated in stress granules, however, mutant MATR3 perturbed stress granule formation as well as cellular viability; the analyzed S85C fibroblasts had a significant decrease in stress granule formation(Aulas et al., 2015).This suggests that while MATR3 does not have a direct role in stress granule development, it may indirectly regulate stress response pathways, possibly through stress granule components.In another study,overexpression and knockdown of endogenous MATR3 in a rat primary neuronal model showed neurotoxic effects as evident from neuronal death, suggesting that a fine balance of MATR3 expression is required for maintaining neuronal homeostasis.Additionally, this cellular model also found that deletion of the ZF2 domain of MATR3 protein arbitrated overexpressionrelated neurotoxicity and that RRM2 deletion induced phase separation of MATR3 into droplets, though no change in toxicity was observed.However, when MATR3- ΔRRM2 was sequestered inside these droplets, survival was increased when compared to MATR3-ΔRRM2 being dispersed in the neurons (Malik et al., 2018).This may imply a neuroprotective nature to formation of the droplets, supporting the theory that overexpressed MATR3 has a harmful gain-of-function.
In the first transgenic MATR3 mouse model,F115C and S85C mutations and human MATR3 wildtype were expressed in the spinal cord and muscles.While S85C expressed poorly in both tissues, F115C mice exhibited significant expression in muscles, but not in spinal cords.By 2 months, wildtype and F115C mice displayed basic muscle pathology, which advanced substantially after 10 months with F115C mice developing partial and full paralysis(Moloney et al., 2018).It should be noted that homozygous MATR3 mice were used in this study, whereas the MATR3 patients harbor autosomal dominant mutations, making it difficult to compare these mice with human patients.A more recent model utilized the CRISPR/Cas9 system to create transgene mice with S85C MATR3 knock-in (Kao et al., 2020).In this study, MATR3 was effectively expressed in the spinal cord and the brain, resulting in neuropathological and behavioral changes such as neuroinflammation in the spinal cord and cerebellum, muscle atrophy, motor impairment,neuromuscular junction defects, and Purkinje cell degeneration.These transgene mice showed similarities to early-stage ALS, helping path the way to future research.
The organismDrosophila melanogasteris a model system useful for the low cost to maintain, many offspring, short lifespan, and simple genetic tracking.TheDrosophilagenome has functional homologs of over 70% of human disease-causing genes.We engineered the firstDrosophilamodels of human MATR3 wildtype,ALS-associated mutations F115C and S85C,and deletion variants in functional domains ΔRRM1, ΔRRM2, ΔZNF1 and ΔZNF2 (Ramesh et al., 2020).Targeted expression of MATR3 inDrosophilamotor neurons and muscles results in muscle degeneration and atrophy,motor defects, and a shortened lifespan,particularly in muscles.Similar results of an age-dependent decline in motor function were seen in transgenic mice expressing the F115C mutant (Moloney et al., 2018).These findings suggest that pathogenic mutations causing a gain-of-function in MATR3 exert their adverse effects through a potential gain of function mechanism.However, the mutations could be working in cis with the functional domains which impacts MATR3 function negatively, because none of the diseaseassociated mutation in MATR3 are found in known functional domains of the protein.To investigate this, transgenic lines were developed with deletion mutations in all functional domains of the protein, which elucidated that RNA-binding domains dictate MATR3 toxicityin vivo.We used our MATR3 fly model to perform a candidate genetic screen, which allowed us to identify rumpestiltskin (rump), a fly homologue of human heterogenous nuclear ribonucleoprotein M (hnRNPM), as a dominant modifier of MATR3 toxicity (Ramesh et al.,2020).hnRNPs are a group of varied RNAbinding proteins implicated in gene regulation,alternative splicing, stress granule formation,cell cycle regulation, and axonal transport(Low et al., 2020).They have also been linked to spinal muscular atrophy, Alzheimer’s disease, ALS, frontotemporal dementia, and other neurological diseases (Low et al., 2020).Intriguingly, hnRNPM functionally and physically interacts with MATR3 in an RNA-dependent fashion in mammalian cells.Further, hnRNPM and MATR3 commonly share RNA targets to perform essential biological processes for the health and survival of neurons.In parallel,another study also identified rump as a genetic modifier of MATR3 toxicity independently (Mori et al., 2013).Rump was shown to substantially ameliorate S85C toxicity when knocked down,as demonstrated in egg-to-adult survival in S85C flies.Rump knock down also substantially increased lifespan in adult S85C flies, but did not have an effect on wildtype flies (Ramesh et al., 2020).This suggests a disease relevant genetic interaction between MATR3 and hnRNPM; combined with rump knock down decreasing S85C insolubility in muscles,this may propose a possible mechanism for mitigating MATR3 toxicityin vivo(Figure 1).
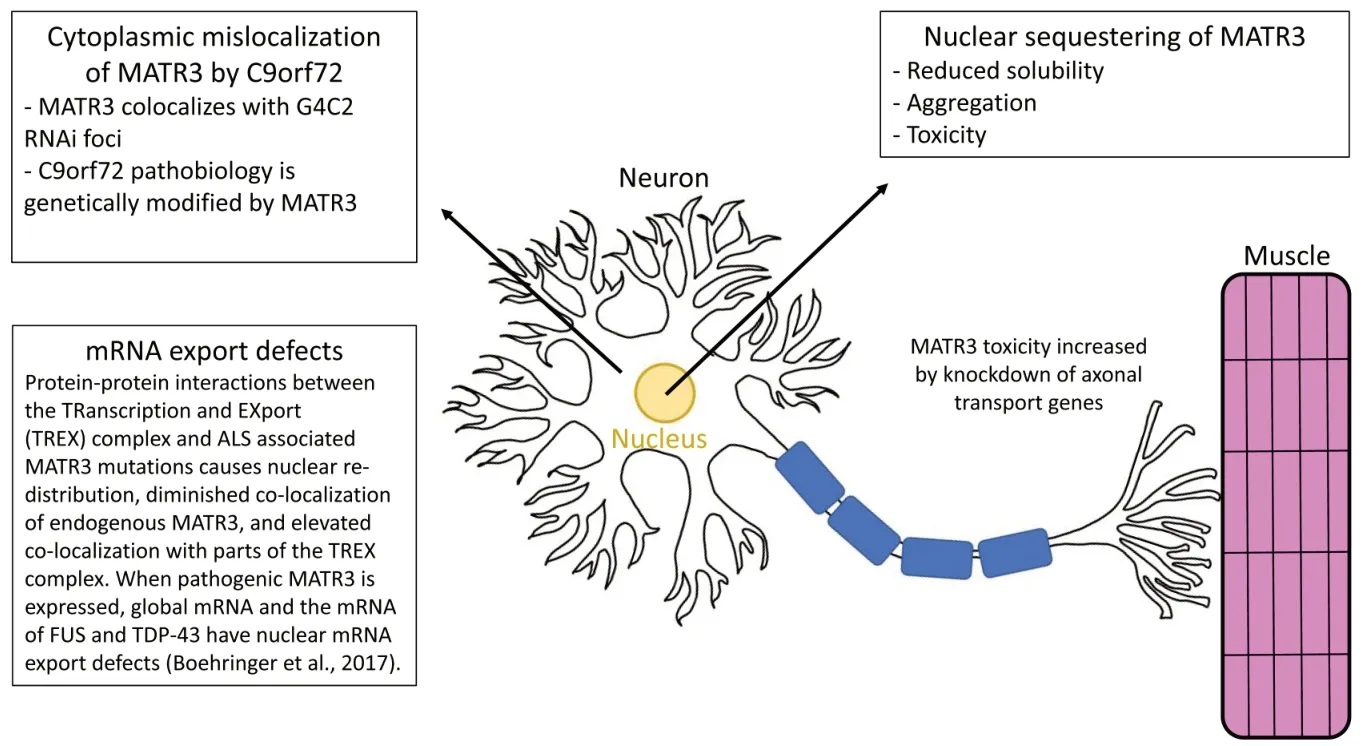
Figure 1| Mutations in MATR3 have been demonstrated to cause ALS, FTD, and distal myopathy by targeting multiple mechanisms.
While the mechanisms behind neuromuscular degeneration caused by MATR3 is largely unknown, it is currently suspected that MATR3 associated ALS and other distal myopathies are a result of multisystem proteinopathy, including dysregulated RNA metabolism; primarily splicing.More specifically, the RNA-binding protein hnRNP, which regulates alternative splicing (Passacantilli et al., 2017), may be a key to determining the mechanism of MATR3, as it was found to be necessary for mutant MATR3 to wield toxicity inDrosophila.These observations prompted us to examine how these two ALScausing genes (C9orf72 and Matrin3) functional interactin vivo; we found that ectopic expression of MATR3 is sufficient to suppress neurodegenerative symptoms associated with expanded C9orf72 (Ramesh et al., 2020).Further, MATR3 positive cytoplasmic inclusions have been identified in an ALS patient known to carry the C9orf72 repeat expansion, suggesting a potential link between these two forms of ALS.MATR3 binds to the G4C2 in C9orf72 and the binding motif recognized by MATR3 has been found to be enriched in sporadic ALS patients’ alternative splicing cassette exons(Prudencio et al., 2015).We have demonstrated in patient neurons and tissues withC9orf72mutation that MATR3 interacts with G4C2 RNA foci, as well as significant alleviation ofin vivoG4C2 associated neurodegeneration by ectopic expression of MATR3, in an RRM-dependent manner (Ramesh et al., 2020).While this characterizes a novel aspect of MATR3 in G4C2 repeat RNA-mediated neurodegeneration,it remains to be seen if MATR3 has a direct effect in C9orf72 associated ALS.However, one possibility is MATR3’s role in regulating C9orf72 G4C2-HRE (hexanucleotide repeat expansion)containing transcripts transcriptionally and/or post-transcriptionally, although more research is needed to substantiate this theory, as well to understand the mechanism of C9orf72 associated ALS in general (Figure 1).
The availability of genetic models is crucial for uncovering mechanisms and genetic modifiers of pathogenic mutations, as well as developing model therapies.TheDrosophilagenetic model has allowed us to make many discoveries of not only MATR3 associated ALS,but several other genetic causes of ALS, as well as other neurological disorders.MATR3 and genetic modifiers have not only been established inDrosophilamodels, but also mammalian models such as mice, helping set the groundwork for prospective primary and preclinical research.The increased availability of genetic models not only opens more avenues for future mechanistic work but is also critical for identifying biological pathways that could be potentially exploited for developing therapeutic interventions.
The present work was supported by the following funding sources: US National Institutes of Health (NIH), National Institute on Neurological Disorders and Stroke (NINDS)and National Institute on Aging (NIA) R01 NS081303, R21 NS094921, R21 NS101661, R21 NS111768, R21 AG064940, R21 NS100055,Muscular Dystrophy Association, the ALS Association, and the Robert Packard Center for ALS at Johns Hopkins (to UBP).
Caroline Ward, Udai Bhan Pandey*
Department of Pediatrics, Division of Child Neurology, Children’s Hospital of Pittsburgh,University of Pittsburgh Medical Center, Pittsburgh,PA, USA (Ward C, Pandey UB)
Department of Human Genetics, School of Public Health, School of Public Health, University of Pittsburgh; Center for Neuroscience Institute,Children’s Hospital of Pittsburgh, University of Pittsburgh Medical Center, Pittsburgh, PA, USA(Pandey UB)
*Correspondence to:Udai Bhan Pandey, PhD,udai@pitt.edu.
https://orcid.org/0000-0002-6267-0179(Udai Bhan Pandey)
Date of submission:March 23, 2021
Date of decision:April 2, 2021
Date of acceptance:May 18, 2021
Date of web publication:August 4, 2021
https://doi.org/10.4103/1673-5374.320986
How to cite this article:Ward C, Pandey UB (2022)Matrin-3 dysfunction in myopathy and motor neuron degeneration.Neural Regen Res 17(3):575-576.
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open accessjournal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Sokol Tod, Wayne State University School of Medicine, USA.

- 中国神经再生研究(英文版)的其它文章
- Pathological mechanisms and therapeutic strategies for Alzheimer’s disease
- Pentadecapeptide BPC 157 and the central nervous system
- OTX2 stimulates adult retinal ganglion cell regeneration
- Mutations in GBA, SNCA, and VPS35 are not associated with Alzheimer’s disease in a Chinese population:a case-control study
- Role of microtubule dynamics in Wallerian degeneration and nerve regeneration after peripheral nerve injury
- Krüppel-like factor 7 attenuates hippocampal neuronal injury after traumatic brain injury