
Novel insights into the pathogenesis of DYT1 dystonia from induced patient-derived neurons
2022-08-15 15:46:18BaojinDing
中国神经再生研究(英文版) 2022年3期
Baojin Ding
Dystonia is a common movement disorder characterized by sustained or intermittent muscle contractions causing abnormal movements and/or postures(Keller Sarmiento and Mencacci, 2021).The dystonic syndromes are classified as primary dystonia (dystonia is the only motor feature without tremor) and the secondary dystonia (dystonia is combined with other movement disorders, such as Parkinsonism).Based on the age of onset, dystonias are also dichotomously classified as childhood onset or adulthood onset.The distribution of affected body parts may change over time and progressively spread of dystonia to previously uninvolved sites.Because the clinical characteristics and underlying causes of dystonia are very heterogeneous,the pathological mechanisms of dystonia remain largely unknown.The diagnosis and etiological definition of this disorder remain challenges.The current therapies, such as anticholinergics, intramuscular botulinum toxin injection and deep brain stimulation,are largely symptom-based and only partially satisfactory (Balint et al., 2018).The childhood-onset torsin dystonia, also called DYT1 dystonia, represents the most frequent and severe form of hereditary primary dystonia, providing an excellent example to understand the pathogenesis of this disease(Gonzalez-Alegre, 2019; Keller Sarmiento and Mencacci, 2021).
Most cases of typical DYT1 dystonia are caused by a heterozygous 3-bp deletion in exon 5 of the TОR1A gene, resulting in the loss of one of the two adjacent glutamate residues (E302 or E303) in the carboxyterminal region of TОR1A protein (ΔE) (Keller Sarmiento and Mencacci, 2021).Protein TОR1A is a member of conserved AAA+(ATPases Associated with diverse cellular Activities) ATPase, which typically use the energy of ATP hydrolysis to disassemble protein complexes in diverse biological processes, such as membrane trafficking,cytoskeleton dynamics, vesicle fusion,response to stress, and the biogenesis of nuclear pore complex (Laudermilch and Schlieker, 2016; Rampello et al., 2020).In humans, TОR1A is widely distributed throughout the central nervous system with high expression in cerebral cortex,striatum, substantia nigra pars compacta,hippocampus, cerebellum and spinal cord,underscoring its critical roles in neural development and functions.At the cellular level, most TОR1A proteins are embedded in the lumen of the endoplasmic reticulum and the endomembrane space of nuclear envelope.The activity of TОR1A depends on the association with one of two transmembrane cofactors, LAP1 (lamina associated polypeptide 1, also named TОR1AIP1) and LULL1 (luminal domain like LAP1, also named TОR1AIP2), suggesting that TОR1A works in a membrane-spanning machinery (Laudermilch and Schlieker, 2016).Although ΔE has been shown to be a loss-offunction mutation, the actual functions of the TОR1A protein in DYT1 dystonia and how it leads to changes in the nervous system to cause dystonia remain poorly understood.While rodent models provide insights into disease mechanisms, significant speciesdependent differences exist because animals with the identical heterozygous mutation fail to show pathology (Gonzalez-Alegre, 2019).However, inaccessibility to patient neurons greatly impedes our understanding of the pathological mechanisms for dystonia.
It is fascinating that reprogramming of human neurons from adult fibroblasts provides an unprecedented approach to deciphering the molecular pathogenesis underlying neurological diseases.Recently,we modeled DYT1 by using human patientspecific cholinergic motor neurons (MNs)that are generated through either direct conversion of patients’ skin fibroblasts(Ding et al., 2020) or differentiation of induced pluripotent stem cells (iPSCs)(Sepehrimanesh and Ding, 2020; Ding,2021).These human MNs naturalistically express the heterozygousTOR1Amutation and display cellular pathology (Ding et al.,2021).Compared to healthy MNs, DYT1 MNs had significantly shorter neurite and many fewer branches, deformed nuclear morphology, fewer nuclear pore complex,and impaired nucleocytoplasmic transport(NCT) that leads to mislocalization of mRNAs and proteins.This is the first demonstration of cellular deficits in a heterozygous state of patient-specific neurons, unravelling important differences between human and mouse models (Ding et al., 2021).
The distinguished features of human DYT1 neurons are deformed nuclear envelope(Figure 1AandB) and dysregulations of nuclear LMNB1 at both expression (Figure 1DandE) and subcellular localization (Figure 1C).Protein LMNB1 is a major component of the nuclear lamina and it contributes to the structural integrity of the nuclear envelope.Оverexpression of LMNB1 has been shown to increase nuclear rigidity.An abnormal extra copy ofLMNB1gene causes autosomal dominant leukodystrophy, which is characterized by loss of myelin in the brain and the spinal cord, leading to movement problems (Kohler et al., 2018).Similarly, I think the increased LMNB1 also contributes to these cellular abnormalities in DYT1 neurons.Consistently, overexpression of LMNB1 indeed disrupts nuclear morphology(Figure 1F).Downregulation of LMNB1 can largely ameliorate all the cellular deficits in DYT1 MNs, including neurodevelopment,nuclear morphology, NCT activities (Figure 1G).These results indicate that dysregulation of LMNB1 may constitute a major molecular mechanism underlying DYT1 pathology.Importantly, this study describes the first cellular model system for human Dystonia using techniques of direct conversation and iPSC system, revealing the great value of disease modeling with human patientderived neurons.
Traditionally, dystonia has been considered a disorder of the basal ganglia, a subcortical nuclei involved in diverse functions including motor control and motor learning.Later studies suggest that dystonia is a network disorder, with involvement of a basal ganglia–cerebello-thalamo-cortical circuit(Balint et al., 2018).Interestingly, recent reports also indicated that the cerebellum and the spinal cord could be other major sites of dysfunction in Tor1a mutant mice(Zhang et al., 2017).The findings that deficits in presynaptic inhibition caused by impaired GABApre neurons in the spinal cord suggest that the abnormalities of proprioceptive and/or muscle spindle feedback contribute to the DYT1 dystonia (Zhang et al., 2017).Using patient-derived MNs, our study further provides evidence that lower MNs are implicated in the pathogenesis of dystonia(Ding et al., 2021).In healthy MNs, nuclear morphology, neurite outgrowth and NCT activities are normal.The majority of mRNAs localize in the cytoplasm (Figure 1H).In DYT1 MNs, upregulated LMNB1 causes thickening of nuclear lamina, thereby leading to abnormal nuclear morphology and impaired NCT, resulting in nuclear accumulation of mRNAs and mislocalized proteins (Figure 1H).Progressive LMNB1 accumulation and subcellular mislocalization are critical pathological features of DYT1 MNs, providing a novel molecular target for therapeutic intervention.
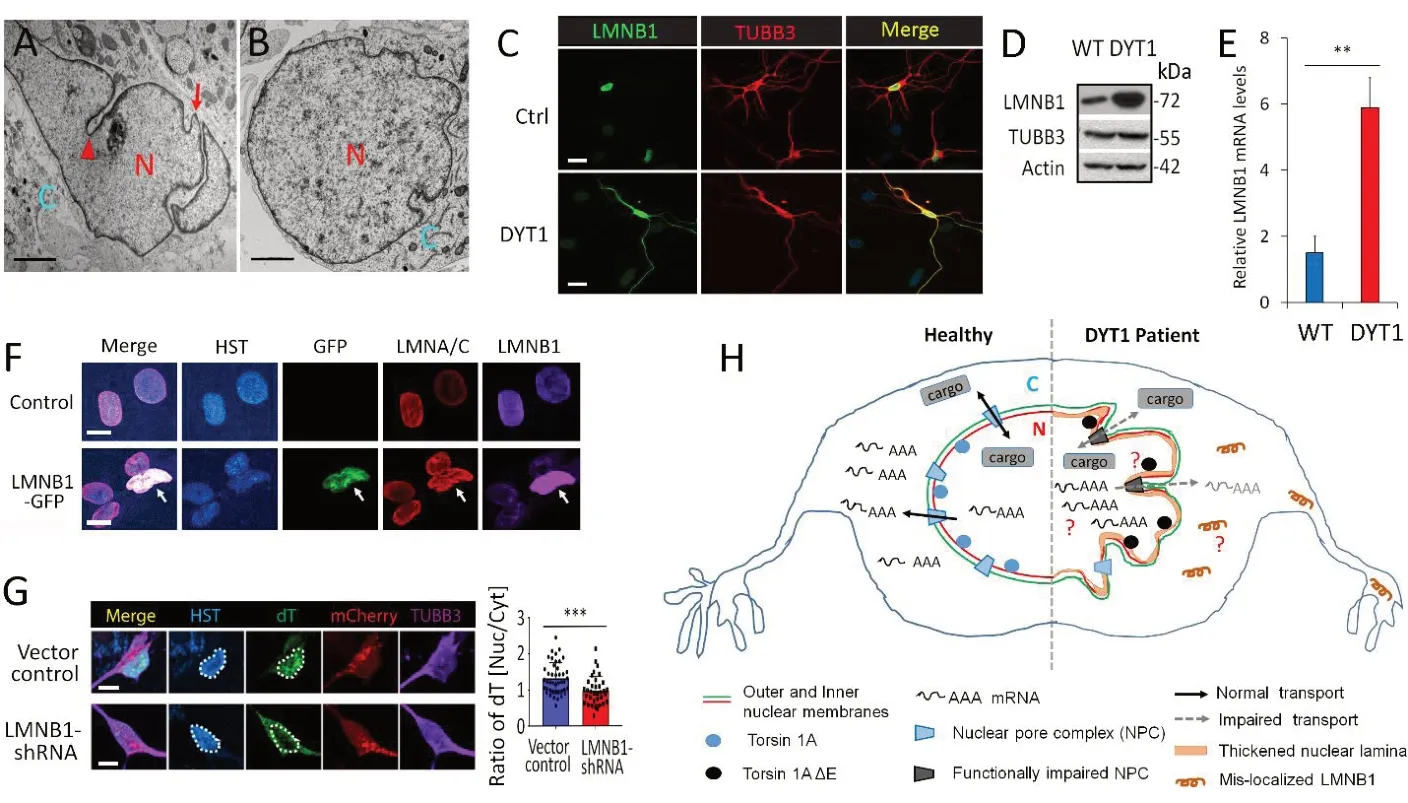
Figure 1|Dysregulation of nuclear LMNB1 in DYT1 patient-derived neurons.
To further understand the dysregulation of LMNB1 underlying the pathogenesis of DYT1 dystonia, the following questions need to be addressed in future studies(Figure 1H).First, to examine the neuronal subtype specificity of LMNB1 dysregulation in DYT1 cells.Dysregulation of LMNB1 has been recapitulated by either ectopic expression of the mutantTOR1Agene or shRNA-mediated downregulation of endogenous TОR1A in healthy control MNs,suggesting that LMNB1 mislocalization are caused byTOR1Ahypoactivity (Ding et al.,2021).In non-MNs that were generated by spontaneous differentiation of DYT1 neural progenitors and differentiation of human SH-SY5Y neuroblasts with ectopic expression of ΔE, no mislocalized LMNB1 can be noticed (Ding et al., 2021), suggesting that LMNB1 mislocalization could be MN-specific.A recent report that neural increase in LMNB1 also contributes to nuclear dysfunction in Huntington’s disease(Alcala-Vida et al., 2021).Оther DYT1 neural subtypes should be examined to test whether TОR1A hypoactivity causes LMNB1 mislocalization in a MN-specific manner.Second, to verify these abnormalities in clinical samples.If these abnormalities identified usingin vitrocellular system also present in clinical samples, these findings will lead to potential therapeutic strategies for DYT1 dystonia.Third, to identify nuclear accumulated transcripts in DYT1 neurons.Impaired nuclear export activity in DYT1 neurons causes the nuclear accumulation of mRNAs (Figure 1H).Are these mRNAs proportional to all transcripts or do they represent some particular subset that are involved in certain neural functions? Do the retained mRNAs share common sequences or secondary structures? These questions could be addressed by biochemical approaches of RNA isolation together with the next-generation sequencing to identify mislocalized transcripts.Fourth, to decipher the molecular mechanisms of how TОR1A ΔE causes dysregulation of LMNB1 and how mislocalized LMNB1 contributes to DYT1 dystonia.Protein co-immunoprecipitation coupled with mass spectrometry analysis could be used to identify ΔE-disrupted factors and mislocalized LMNB1-interacting proteins in DYT1 MNs.The abnormal protein-protein interactions and/or disrupted signaling pathways could provide novel insights into the dysfunctions of DYT1 neurons.No matter RNAseq assay or co-immunoprecipitation coupled with mass spectrometry analysis,generation of patient-specific MNs with high purity and yield is absolutely required.Excitingly, we have developed a protocol by which highly pure patient-specific MNs could be generated from hiPSC (Sepehrimanesh and Ding, 2020; Ding, 2021), providing a solid foundation to address these questions using biochemical approaches.
This work was supported by National Institute of Neurological Diseases and Stroke, No.NIH/NINDS NS112910 (to BD) and Department of Defense (DoD) Peer Reviewed Medical Research Program (PRMRP) Discovery Award,No.W81XWH2010186 (to BD).
Baojin Ding*
Department of Biology, University of Louisiana at Lafayette, Lafayette, LA, USA
*Correspondence to:Baojin Ding, MD, PhD,Baojin.Ding@Louisiana.edu.
https://orcid.org/0000-0002-2149-2599(Baojin Ding)
Date of submission:March 21, 2021
Date of decision:April 23, 2021
Date of acceptance:May 26, 2021
Date of web publication:August 4, 2021
https://doi.org/10.4103/1673-5374.320978
How to cite this article:Ding B (2022) Novel insights into the pathogenesis of DYT1 dystonia from induced patient-derived neurons.Neural Regen Res 17(3):561-562.
Copyright license agreement:The Copyright License Agreement has been signed by the author before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Carlo Colosimo, Santa Maria Hospital of Terni, Italy.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Pathological mechanisms and therapeutic strategies for Alzheimer’s disease
- Pentadecapeptide BPC 157 and the central nervous system
- OTX2 stimulates adult retinal ganglion cell regeneration
- Mutations in GBA, SNCA, and VPS35 are not associated with Alzheimer’s disease in a Chinese population:a case-control study
- Role of microtubule dynamics in Wallerian degeneration and nerve regeneration after peripheral nerve injury
- Krüppel-like factor 7 attenuates hippocampal neuronal injury after traumatic brain injury