
HPLC法测定扶正化瘀片中五味子醇甲、乙含量
2022-07-30 07:07:30张玉杰谢富玲付庆帅李荣胜潘一峰
上海医药 2022年13期
张玉杰 谢富玲 付庆帅 李荣胜 潘一峰,2
(1. 上海黄海制药有限责任公司 上海 200942;2. 上海现代中医药股份有限公司 上海 200050)
扶正化瘀片是由五味子、桃仁、绞股蓝、丹参、发酵虫草菌粉和松花粉六味药材组成的复方中成药。具有活血祛瘀、益精养肝的功效,用于乙型肝炎肝纤维化属瘀血阻络、肝肾不足证者[1]。五味子作为使药,含有丰富的木脂素成分,其中五味子醇甲、乙是主要的药效成分[2-5]。目前《中国药典》2020 年版标准五味子仅以五味子醇甲作为含量控制成分[6],而USP 41/NF36 在此基础上增加了五味子醇乙等4 个成分的含量控制[7]。扶正化瘀片质量标准中仅以五味子甲素的TLC 的鉴别作为判断依据,很难准确地表征木脂素成分的含量。同时扶正化瘀片成分复杂,很多成分极性相近或属同分异构体,其中戈米辛D、J 出峰位置与五味子醇甲、乙也十分接近,采用已报道的HPLC 方法[8]流动相体系测定扶正化瘀片中五味子醇甲、乙的含量存在分离度和积分值重现性差,甚至目标峰和其他成分完全重合为共流峰的现象,从而影响准确定量。另外刘珊等[9]、邢心睿等[10]采用UPLCMS/MS 和UHPLC-Q-TOF/MS 技术测定人体口服扶正化瘀片后在血浆中检测到以原型化合物入血成分有五味子醇甲、乙等多种化合物,得到较明显的血药浓度与时间曲线及药代动力学参数。因此建立适合扶正化瘀片中五味子醇甲、乙含量测定方法有助于扶正化瘀制剂质量标准的提升,为药理、毒理研究提供可靠的基础数据。
1 材料和方法
1.1 材料
1)仪器 Agilent 1260ⅠⅠ高效液相色谱仪、Agilent Zorbax Extend C18柱(4.6 mm × 250 mm, 5 μm)[安捷伦科技(中国)有限公司];XP-105 电子天平[梅特勒-托利多国际贸易(上海)有限公司];HWS-28 恒温水浴锅(上海一恒科技有限公司);SK7200 LHC 超声清洗仪(上海科导超声仪器有限公司)。
2)试剂与药物 五味子醇甲(批号110857-201815,纯度99.7%,中国食品药品检定研究院);五味子醇乙(批号3249,纯度98.0%,上海诗丹德标准技术服务有限公司);扶正化瘀片0.8 g(批号S200404,上海黄海制药有限责任公司);甲醇为HPLC 级(上海星可高纯试剂有限公司);纯水自制。
1.2 方法
1.2.1 色谱条件
采用Agilent Zorbax Extend C18色谱柱;流动相为甲醇(A)-水(B),梯度洗脱(0~55 min,55% A;55~57 min,55%~90% A;57~60 min,90% A;60~62 min,90%~55% A;62~65 min,55% A);流速1.0 mL/min;检测波长250 nm;柱温30 ℃;进样量20 μL。
1.2.2 对照品溶液制备
精密称取对照品五味子醇甲24.00 mg、五味子醇乙16.00 mg,分别置10 mL 量瓶中,加70%甲醇溶解,定容,摇匀,即得质量浓度分别为2.39、1.57 mg/mL 对照品储备液,于4 ℃保存备用。
1.2.3 供试品溶液的制备
取本品10 片称重,研细,混匀,取约1.5 g,精密称定,置于具塞锥形瓶中,精密加入70%甲醇20 mL,密塞,称定重量,超声处理15 min,放冷,再称定重量,加70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
1.2.4 专属性实验
取供试品、混合对照品溶液、阴性供试品溶液(除去五味子提取物)以及五味子提取物供试品溶液,进行色谱测定。
1.2.5 线性实验
分别精密吸取2 种对照品溶液适量,置于不同规格容量瓶中,加70%甲醇稀释至刻度,配制成6 个不同浓度的混标溶液,进行色谱测定。
1.2.6 精密度实验
在不同日期,采用不同仪器,由两位实验员平行制备6 份供试品溶液,进行色谱测定。
1.2.7 准确度实验
分别精密称取本品0.75 g,置具塞锥形瓶中,精密加入上述对照品混标储备液5、10、15 mL,精密加入15、10、5 mL 70%甲醇,摇匀,称重,超声处理15 min,放冷,再称重,加70%甲醇补足减失的重量,摇匀,滤过,取续滤液,平行制备3 份即得低、中、高浓度准确度测定溶液,进行色谱测定。
1.2.8 稳定性实验
取同一供试品溶液和同一对照品溶液,于25 ℃下分别存放0、6、12、24、48 h,进行色谱测定。
2 结果
2.1 专属性
样品中五味子醇甲、乙色谱峰与其他峰能达到基线分离。空白溶液与阴性供试品溶液在对照品保留时间处无干扰峰,供试品溶液在与对照品溶液相同保留时间位置出峰,且待测成分与邻近干扰峰分离度大于1.5,表明该方法专属性良好(图1)。

图1 HPLC色谱图
2.2 线性关系考察
以对照品质量浓度为横坐标(x),峰面积为纵坐标(y)进行线性回归,五味子醇甲:y=39 974.00x-58.30,r=0.999 9,线性范围0.024 0~1.203 4 mg/mL;五味子醇乙:y=33 093.60x-126.43,r=0.999 9,线性范围0.015 7~0.784 5 mg/mL,两成分在各自浓度范围内呈现良好的线性关系。
2.3 精密度(重复性、中间精密度)
实验员1 测定平行6 份样品中五味子醇甲、乙的平均含量分别为0.206%(RSD 0.59%)、0.052%(RSD 1.94%);实验员2 测定平行6 份样品中五味子醇甲、乙的平均含量分别为0.205%(RSD 0.43%)、0.053%(RSD 0.95%);表明精密度良好。
2.4 准确度
低、中、高水平样品中五味子醇甲的平均加样回收率为105.63%(RSD 0.90%,n=9);五味子醇乙平均加样回收率为103.24%(RSD 3.81%,n=9),表明方法准确度良好(表1)。
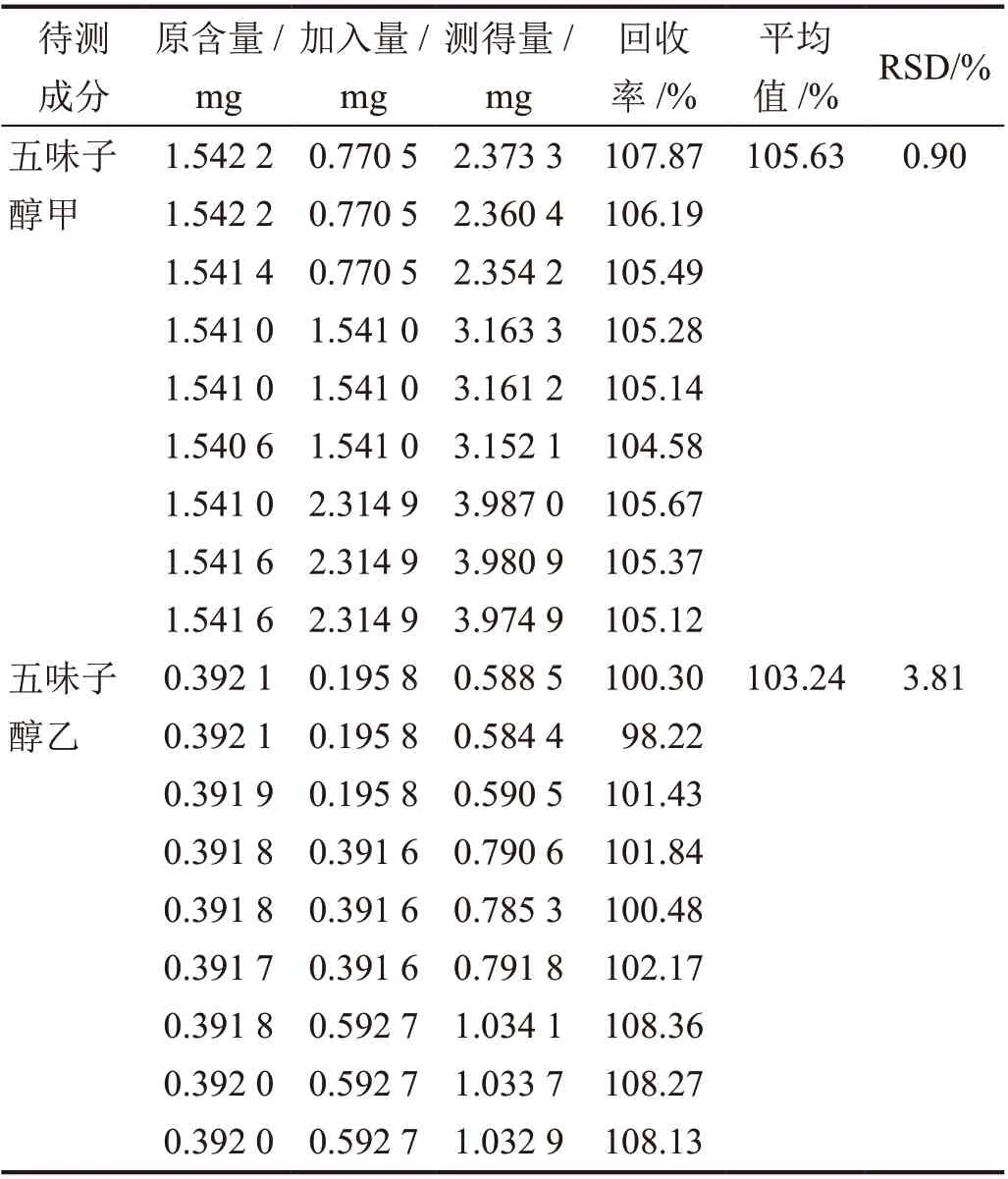
表1 准确度实验结果
2.5 稳定性试验
供试品及对照品溶液中五味子醇甲、乙的峰面积RSD 分别为0.49%、1.84%;0.68%、0.95%。表明对照品与供试品溶液中待测成分在48 h 内稳定性良好。
2.6 含量测定
根据建立的方法对10 批次不同批号的扶正化瘀片进行五味子醇甲、乙含量测定,被测组分色谱峰与其他峰能达到基线分离,峰形尖锐对称、重现性良好,五味子醇甲、乙含量范围分别在0.093%~0.181%、0.025%~0.056%(表2)。
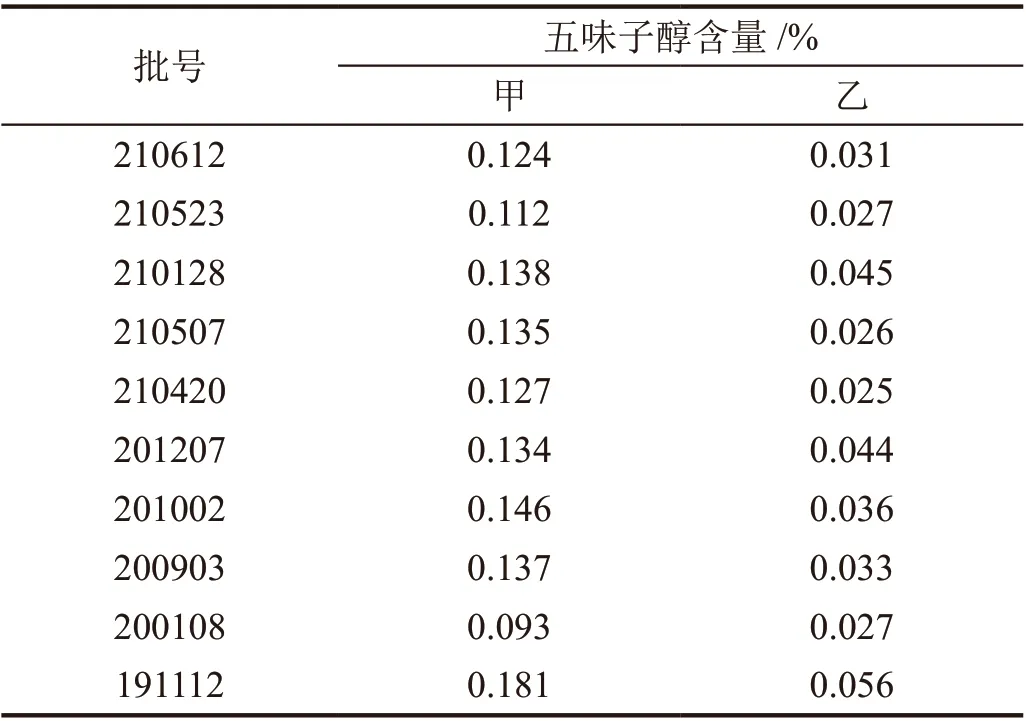
表2 扶正化瘀片中五味子醇甲、乙含量
3 讨论
在色谱条件优化研究中,参考陈立柱等[8]、刘晶等[11]、支旭然等[12]、王艳等[13]已发表的文献对乙腈-水、甲醇-水流动相体系调整比例后均进行了考察,发现不同流动相体系下,均能达到目标化合物的有效分离,但是甲醇-水体系下五味子醇甲、乙的出峰时间更短,出于缩短分析时间、节约成本的角度,选择甲醇-水作为色谱测定的流动相体系。
对五味子醇甲、乙对照品溶液进行全波长(200 ~400 nm)扫描,五味子醇甲、乙分别于210、250 nm 处有最大吸收,250 nm 波长下基线平稳、目标峰周围干扰峰少且响应低,结合《中国药典》2020 年版及相关文献,最终选择250 nm 为检测波长。
考察了流动相中添加剂(0.1%甲酸、0.1%磷酸、2 mmol/L 乙酸铵)对五味子醇甲、乙的色谱峰面积、理论塔板数、分离度、拖尾因子的影响。研究表明,以磷酸为添加剂(浓度0.1%)时,五味子醇乙峰与杂质峰(戈米辛J)分离度不佳,其余添加剂对分析结果无显著性影响,结合目标化合物本身性质,选择不添加任何添加剂,以水-甲醇体系作为流动相按一定的比例梯度洗脱。
考察了不同柱温(20、25、30、35 ℃)对五味子醇甲、乙的色谱峰面积及相关参数的影响。结果表明,各柱温对分析结果无明显影响,为便于控制柱温,选择柱温为30 ℃。
在准确度考察研究中,高浓度下五味子醇乙测得的回收率略高,其原因是由于五味子醇乙含量较低。参照《中国药典》2020 年版四部< 9101>分析方法验证指导原则,其回收率为85%~110%,仍符合相关要求。
综上所述,本研究建立了HPLC 方法测定扶正化瘀片中五味子醇甲、乙含量,有助于扶正化瘀片及相关剂型的质量控制,可为质量标准的提升、药理及毒理研究提供参考依据。该分析方法专属性强、准确度高、耐用性良好、操作简便且符合方法验证相关要求。
猜你喜欢
Journal of Traditional Chinese Medicine(2023年1期)2023-02-15 13:44:58
基层中医药(2022年1期)2022-07-22 07:21:36
Journal of Traditional Chinese Medicine(2022年3期)2022-07-20 15:54:08
Journal of Traditional Chinese Medicine(2022年3期)2022-07-20 15:53:38
中成药(2018年12期)2018-12-29 12:26:02
中成药(2018年7期)2018-08-04 06:04:04
养生保健指南(2018年3期)2018-04-13 09:21:02
中成药(2017年3期)2017-05-17 06:09:06
中成药(2017年3期)2017-05-17 06:09:01
药学研究(2015年11期)2015-12-19 11:10:54
