
差分电荷密度在电子结构分析中的教学实践
2022-07-30 03:22章佳菲白鸽徐余幸吴文清刘亚滕波涛
大学化学 2022年6期
章佳菲,白鸽,徐余幸,吴文清,*,刘亚,滕波涛,2,*
1浙江师范大学化学与生命科学学院,浙江 金华 321004
2天津科技大学化工与材料学院,天津 300457
基于第一性原理的理论计算可以从分子、原子水平研究物质的结构,并从微观电子角度研究原子、分子、团簇、固体材料等的电子结构,探究材料的结构-性能间的构效关系,为材料的改性及新材料的开发提供理论支撑。以催化材料研究与开发为例,通过理论计算催化剂的电子结构及反应物与催化剂间相互作用,认识催化反应的活性中心与活性物种,进而研究催化反应机理,建立可能的催化剂结构与性能的关联,为催化剂的理性设计提供重要依据[1]。随着计算化学与高性能计算机的迅速发展,基于第一性原理的理论计算在化学与材料研究中广泛应用,很多高校为研究生及高年级本科生开设了相关的课程[2-4],其中电子结构分析是教学的重点之一。
在众多电子结构分析方法中,电荷可以获得体系中每个原子相比孤立原子的电子得失情况;静电势可反映体系不同位置的亲电与亲核性;电荷密度反映了体系中电子分布情况;能带结构与态密度可以获取材料在价带、导带等的电子结构;分子轨道可以获取电子在分子不同能级轨道中的分布。与以上电子结构分析方法不同,差分电荷密度可以研究分子、团簇、固体材料、分子与固体材料间相互作用导致的电荷重新分布,因此广泛应用于电子结构分析[5]。作者在长期的理论计算研究与“密度泛函理论在化学中的应用”课程教学过程中发现,由于差分电荷密度计算要根据研究对象和研究目标的不同,具体计算过程不同,计算的结果也大不相同。如果不能正确理解其概念,选择合适的计算方法,则无法获取正确的电子结构信息。因此,差分电荷密度计算是电子结构分析教学的难点和重点。本文系统整理了常见与特殊差分电荷密度计算的类型、计算方法与适用研究体系,通过课堂教师讲解与演示、学生练习,结合探究性课后作业,使学生深入理解、掌握这一重要电子分析方法的概念、目标与计算方法,为相应教学与科研提供重要信息。
1 差分电荷密度定义、计算与讨论
差分电荷密度主要考查研究对象在相互作用中电子的移动与再分布情况,按照差分电荷密度计算方法的不同,差分电荷密度可以分为原子基、碎片基与特定对象的差分电荷密度以及自旋电荷密度。计算差分电荷密度的程序有很多,本文给出的例子均是采用Vienna ab initio simulation package (VASP)计算的,差分电荷密度图由VESTA软件获得。下面分别对不同类型的差分电荷密度进行讨论分析。
1.1 原子基差分电荷密度
原子基差分电荷密度是指研究对象(分子、团簇或模型催化剂)成键后的电子云密度与组成其结构的原子电子云密度之差。例如,分子AB由原子A与B组成,其差分电荷密度为分子AB的电子云密度分布减去原子A与B的电子云密度,计算公式如下:

其中ρAB、ρA与ρB分别为分子AB、原子A与B的电子云密度。若研究对象为多原子分子、团簇或模型催化剂,其差分电荷密度为系统的总电子云密度减去组成其结构的各原子电子云密度之和,计算公式为:
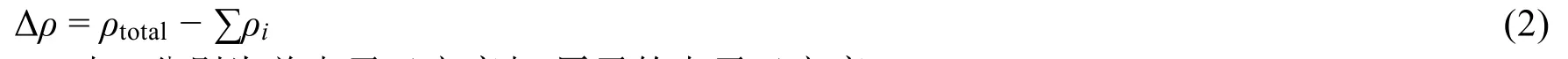
其中ρtotal与ρi分别为总电子云密度与i原子的电子云密度。
原子基差分电荷密度可得到研究对象(分子、团簇或催化剂)成键和成键电子耦合过程中的电子移动与成键极化方向等性质,多用于分析分子、团簇成键情况。
图1分别给出了CO2、O2、四层Pd(111)表面原子基差分电荷密度图。由图1(a)可以看出,当C与O原子相互作用形成CO2时,电子发生了重新排布,C原子以失电子为主(蓝色),O原子以得电子为主(黄色),电子在C与O原子间重新分布形成了C―O共价键;同时CO2分子得失电子情况上下对称,是非极性分子。O2为同核双原子分子,O原子在得失电子的同时,O―O间形成较强相互作用的共价键,如图1(b)所示。同时由于同核双原子分子对称,O―O键没有极性,是非极性分子。由图1(c)的Pt(111)四层周期性模型的原子基差分电荷密度图可以看出,电子在Pt(111)模型的上下表面略有富集,而中间两层以失电子为主,这一结果也可由相应的电荷分析进一步证明。
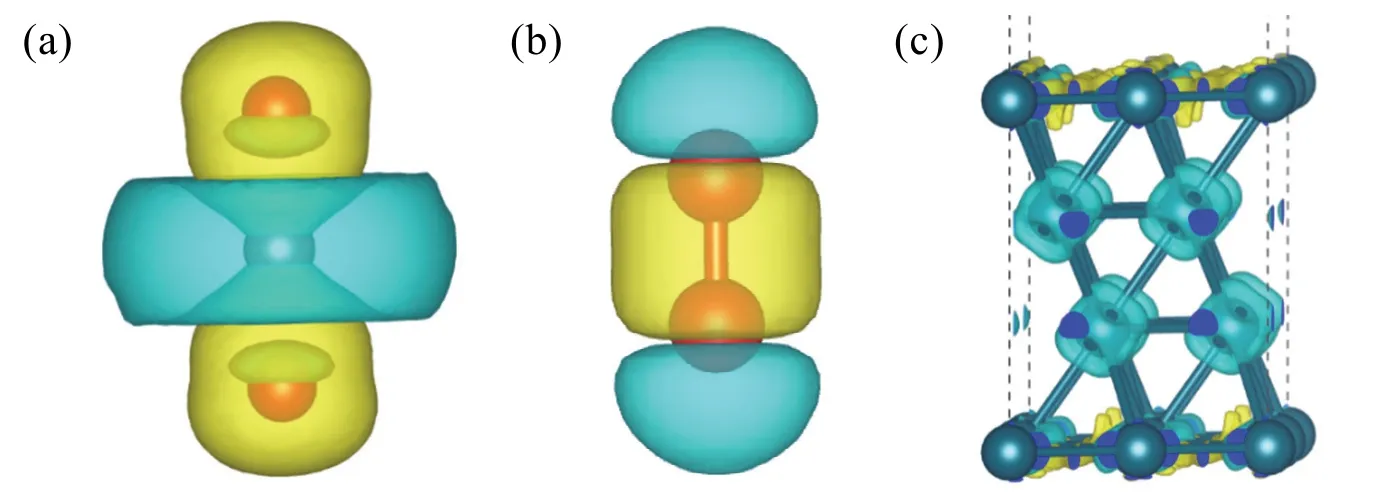
图1 CO2 (a)、O2 (b)与Pd(111) (c)的原子基差分电荷密度图
1.2 碎片基差分电荷密度
碎片基差分电荷密度定义:体系总电荷密度减去吸附分子与基底(或模型催化剂)的电荷密度,计算公式如下:

其中ρA/S为吸附分子与基底的总电荷密度,ρA和ρS分别为吸附分子与基底(或模型催化剂)的电荷密度。通过碎片基差分电荷密度的计算和分析,可以分析吸附分子与基底(或模型催化剂)间的电子相互作用。
图2给出了CF3CH2I在Ag(111)表面吸附时CF3CH2I与接触的Ag原子相互作用的差分电荷密度图。由图中可以看出,当等值面取值为0.01 e·a0-3时,几乎看不到两者的电子相互作用;当等值面取0.001 e·a0-3时,两者电子相互作用比较明显,即Ag失电子(蓝色),CF3CH2I的最高占据轨道(HOMO)nI轨道(未成键p电子)失电子,其最低未占据轨道(LUMO)σC―I*得电子(黄色),Ag与I原子之间形成一定的相互作用,如图2(b)中Ag―I之间的黄色部分所示。差分电荷密度直观清晰地表明了吸附分子与模型催化剂Ag(111)表面的电子相互作用。从差分电荷密度不同的等值面也可以看出,CF3CH2I与Ag有一定相互作用,但不是很强,这与其计算的吸附能为-0.27 eV相一致[6]。
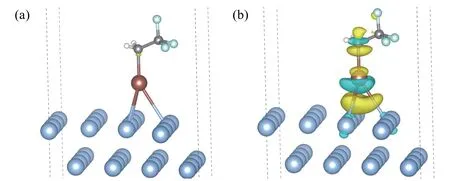
图2 CF3CH2I/Ag(111)表面的差分电荷密度图
与CF3CH2I不同,当CF3CH2与Ag(111)相互作用时,由图3不同等值面的差分电荷密度图可以看出,当等值面取0.01 e·a0-3时,两者电子相互作用就很明显,Ag失电子,CH2CF3得电子;当等值面取0.001 e·a0-3时,两者强电子作用更明显,这与其较强的吸附能(-1.57 eV)相一致。
图3 CF3CH2/Ag(111)表面的差分电荷密度图
基于碎片基的差分电荷密度还可研究金属纳米粒子与氧化物载体间的电子转移与分布情况。图4分别给出了金团簇Au3、Au9与CeO2(111)表面相互作用时的电子转移情况。由图4可以看出,当Au3与Au9团簇与氧化铈相互作用时,金团簇中与氧化铈载体接触的金原子失电子(金原子中蓝色部分),氧化铈载体中的Ce4+离子得电子,形成Ce3+(铈离子中黄色部分)。同时还可以发现,Au9团簇与氧化铈载体相互作用,主要是金团簇底部与氧化铈接触的原子失电子[7]。因此,差分电荷密度可以直观地得到金属纳米粒子与氧化物载体相互作用产生的电子转移与分布。

图4 Au3 (a)、Au9 (b)与CeO2(111)表面相互作用的差分电荷密度图
1.3 自旋电荷密度
自旋电荷密度是特殊的差分电荷密度,是体系自旋向上的电荷密度减去自旋向下电荷密度,具体计算公式如下:

其中ρup与ρdown分别为自旋向上与自旋向下的电子云密度。
自旋电荷密度通常用来研究体系(分子、团簇、模型催化剂)的自旋性质[8]。以催化氧化反应中非常关注的氧气、超氧、过氧等氧物种为例,由于O2分子的最稳定结构为三重态,分子中两个自旋单电子分别填充于2个2π*轨道,其自旋电荷密度图为哑铃形,如图5(b)上方图所示;当其得到一个电子形成超氧物种O2-,只有一个自旋电子在2π*轨道,如图5(a)所示,其自旋电荷密度图为蝴蝶形,如图5(b)中间图所示;当其得到两个电子形成超氧物种O22-,2π*没有自旋电子,自旋电荷密度图看不到自旋电子,如图5(b)下方图所示。因此,可以根据图5(b)中的自旋电荷密度图的分子氧、超氧与过氧的自旋电子结构作为不同氧物种的识别依据。
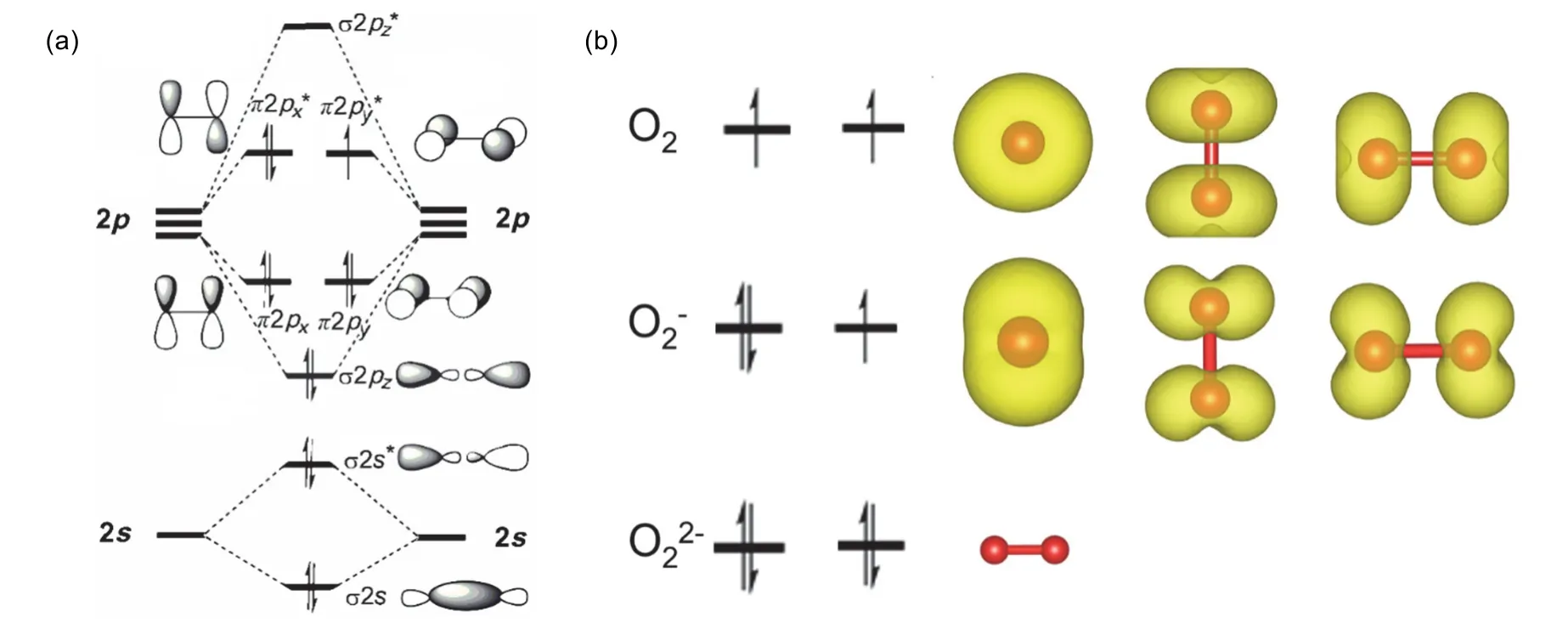
图5 不同氧物种电子排布与自旋电荷密度图
图4的碎片基的差分电荷密度图结果表明,金团簇负载在氧化铈载体上,由于金团簇与载体相互作用产生的电子转移金团簇失电子,氧化铈中的Ce离子得电子。但是,由于碎片基差分电荷密度图中显示金团簇与氧化铈各有电子得失,Ce离子得电子情况并不直观。由于Ce原子外层电子排布为5s25p64f15d16s2,当其为Ce4+离子时,没有自旋电子,当其得一个电子生成Ce3+离子时,在f轨道填充一个自旋电子,因此在自旋密度图中表现出f轨道特有的轨道形状。图6所示的Aux/CeO2(111)的自旋电荷密度图可以清晰地看到,Au3/CeO2(111)中Au3与氧化铈相互作用,Au3上的电子转移到一个Ce4+上,形成一个Ce3+离子,而Au9/CeO2(111)由于有较多Au原子与载体相互作用,导致3个电子转移到氧化铈载体上,形成3个Ce3+离子。
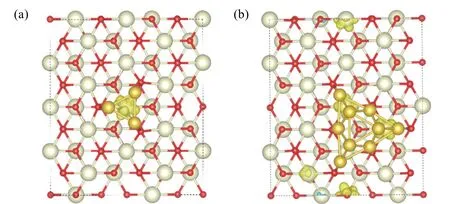
图6 Au3/CeO2(111) (a)与Au9/CeO2(111) (b)的自旋电荷密度图
自旋电荷密度还可以研究吸附物种与载体相互作用发生的电子转移。如图7(b)所示,当氧化铈载体表面失去一个氧原子形成一个氧缺陷时,在氧化铈载体表面留下两个电子,被两个Ce4+离子获取形成两个Ce3+离子,可以由图7(b)看出载体表面有两个4f轨道的自旋电子。当O2分子吸附于其中一个Ce3+离子上,其中4f轨道上的1个电子转移至O2上形成时O2-,具有超氧自旋电荷密度图的特征,同时Ce3+离子失电子再次变成Ce4+,如图7(a)所示。当O2分子吸附于表面氧缺陷时,载体上两个4f轨道自旋电子都转移至O2上形成时O22-,载体与超氧均没有自旋电子,即两个Ce3+离子均被氧化为Ce4+,如图7(c)所示。因此,自旋电荷密度图清晰、直观地给出了表面吸附物种与载体相互作用产生的电子转移情况[9]。
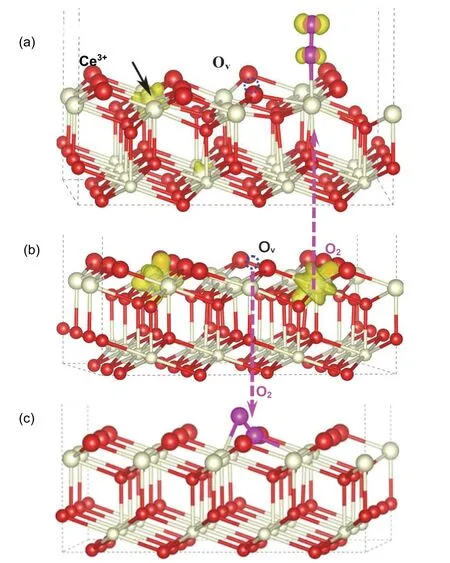
图7 CeO2(111)表面超氧与过氧物种形成过程
1.4 特定对象的差分电荷密度
特定对象的差分电荷密度研究是根据研究体系与目标的不同,无法由原子基或碎片基进行差分电荷密度的计算,需要根据研究体系的特点及研究目标不同,选择特定的电荷密度差分背景,其计算公式可以用下式表示:

其中ρA与ρB分别为研究对象与特定差分对象的电荷密度。
众所周知,由于金属团簇面、棱、角上的原子配位不饱和度较高,可以形成电子富集,导致其电荷密度较内部原子高。如图8(a)为Pd35团簇的电荷密度图,由于团簇内部金属与表面和棱角上的电子云密度相差不是特别大,从团簇的电荷密度图8(a)中很难看出电子在面、棱及角上发生富集。图8(b)为基于原子基的差分电荷密度图,其计算方式为团簇的电荷密度减去各孤立Pd金属原子的电荷密度。因此,在部分观察到电子云在团簇的角、棱富集的同时,主要体现了电子云在两个相邻Pd原子间的富集,表明Pd金属间形成了金属键。
为清晰获取电子在Pd35团簇的面、棱与角上的富集,可以将体相Pd金属形成的电荷密度作为背景,用Pd团簇的电荷密度减去体相Pd金属的电荷密度,如图8(c)所示。其中,体相Pd金属的电荷密度由相同格子大小的体相Pd金属超晶胞计算。图8(c)可以看出,电子在金属团簇的面、棱、角的富集程度依次增加,同时图8(c)中观察不到金属间相互作用形成的金属键电子,因此可以清晰、直观地展示金属团簇电子云富集情况,为解释金属催化剂面、棱、角不同反应性能提供了一定的理论依据。
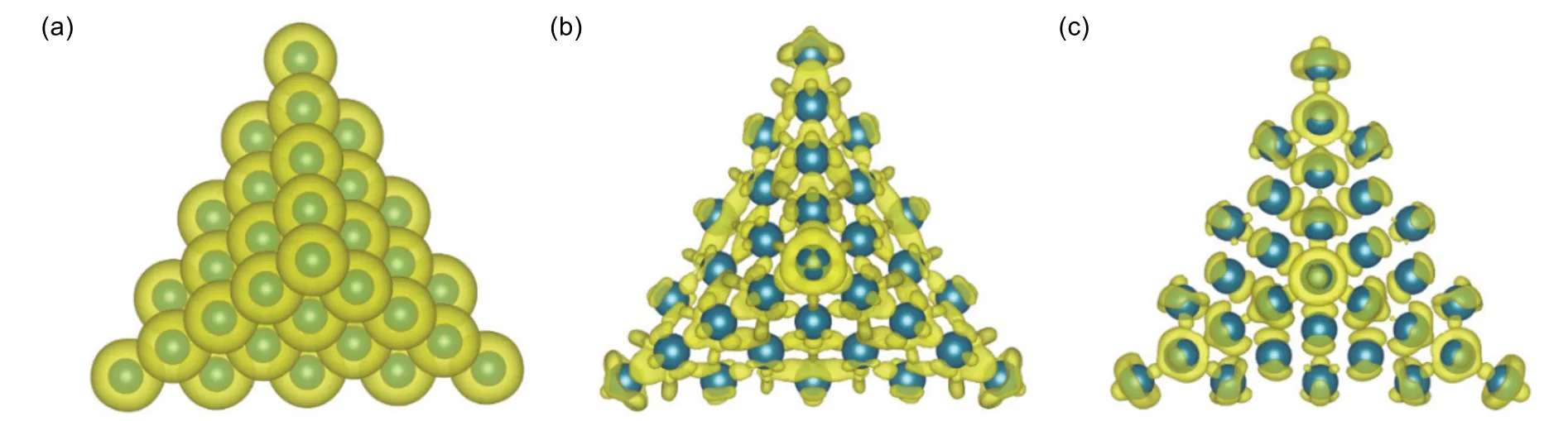
图8 Pd35团簇的电荷密度图(a)、基于原子(b)与体相金属(c)电子分布的差分电荷密度图
综上,通过教师课堂的系统讲解与分析,使学生掌握以下四点:(1) 原子基差分电荷密度是以各孤立原子自身电荷密度的和为差分背景的,可得到研究对象成键和成键电子耦合过程中的电子移动与成键极化方向等性质;(2) 碎片基的差分电荷密度以吸附分子与碎片的电荷密度为差分背景,可获得吸附物种与吸附剂间的电子转移与分布情况,即电子转移主要发生在哪些原子之间,转移电子的数量,进而初步判断出两者的相互作用强弱;(3) 自旋电荷密度是体系自旋向上的电荷密度减去自旋向下电荷密度,可获得磁性体系中自旋电子分布、传递及特征物种识别等信息;(4) 特定对象的差分电荷密度是根据具体的研究目标,选择特定的电荷密度背景差分,获取差分电荷密度信息,进而分析电子结构,为解释特定化学结构、反应性能等提供微观电子解释。
2 结语
通过课堂教师对基本概念的讲解,简单实例的计算、作图演示与学生的当堂练习,激发了学生的学习兴趣,加深了学生对概念的理解。结合课后探究性作业,学生能够深入理解原子基与碎片基差分电荷密度、自旋电荷密度及特定对象的差分电荷密度的概念,并与结构化学中的原子、双原子分子、多原子分子的结构和性质,配位化合物、金属的结构和性质相关知识关联,初步掌握根据不同研究目标,选择合适计算方法,开展相应计算,获取电子结构信息,助力其科研工作;同时培养学生具体问题具体分析,进而解决问题的能力。
猜你喜欢
数学杂志(2022年5期)2022-12-02
湘潭大学自然科学学报(2022年2期)2022-07-28
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化·中考版(2021年10期)2021-11-22
新世纪智能(数学备考)(2021年5期)2021-07-28
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
新高考·高一物理(2015年6期)2015-09-28
新高考·高一物理(2015年6期)2015-09-28
