
独立存在的π单键
2022-07-30 03:22刘丹岳立张利李忠曙
大学化学 2022年6期
刘丹,岳立,张利,李忠曙
中山大学化学学院,广州 510006
化学是一门重要的基础课。化学教学不仅传授专业基础知识和基本理论,还应该注重引导学生了解前沿新知识、新理论和新方法。这要求相应的一线教师紧跟科学前沿,从而实现对教师业务能力的不断提高。在目前主流的化学教科书中,所涉及到的共价键有两种,一种是头碰头的σ键,一种是肩并肩的π键。值得注意的是,所有的单键都是σ键,双键或者三键则由一个σ键和一个或两个π键组成。由此可见肩并肩的π键都是伴随着σ键而存在,没有单独存在。爱思考的同学会问起,是否有单独存在的π单键呢?如果有的话,会是在什么样的化合物中呢?本文将首先从理论上分析π单键能否独立存在,随后综述了近年来报道的拥有独立存在的π单键的化合物的设计与合成,这两方面的内容都证实了π键是可以独立存在的,最后详细分析了这些独立存在的π单键的电子结构特点,为新型独立π单键化合物的设计合成提供参考。这些内容的讲解体现了前沿的科研成果并不全是深奥难懂的,且与基础教学内容环环相扣,可作为教学内容的补充知识点,引导学生探讨问题,从而增加学习兴趣。
1 σ键和π键的独立性分析
分子的电子结构决定了它们的物理化学性质,包括反应活性、氧化还原性、光学性质等,而这些性质往往都来源于分子中前线轨道上电子的变化。分子的前线轨道主要是指最高占据分子轨道(Highest Occupied Molecular Orbital,HOMO)和最低未占分子轨道(Lowest Unoccupied Molecular Orbital,LUMO)。例如,在乙烯分子中,C=C双键由一个σ键和一个π键组成,后者由于交叠程度低,导致其键强比σ键弱,因此乙烯分子的反应性主要体现在π键上。而从分子轨道角度来看,两个原子轨道(p轨道)相态一样时相互叠加形成π成键,相态相反时相互排斥形成π反键,分别为乙烯分子的HOMO和LUMO (图1a,为了便于对比分析,除非用到文献中的数据,本文中所有化合物的计算结果均为作者在M062X/def2TZVP等级上计算得到)。
虽然目前教科书上所描述的π键总是伴随着σ键而存在,但从成键模式来看,形成π键的p轨道和形成σ键的杂化轨道之间无相互依存性,因此在图1b中可以看出,若将sp2杂化的两个碳中心左右交换,中间两对杂化轨道之间由其他原子或原子团相连接,在两个碳中心距离合适时,平行的p轨道之间即能形成独立的π单键。那么,这样环状结构存在吗?
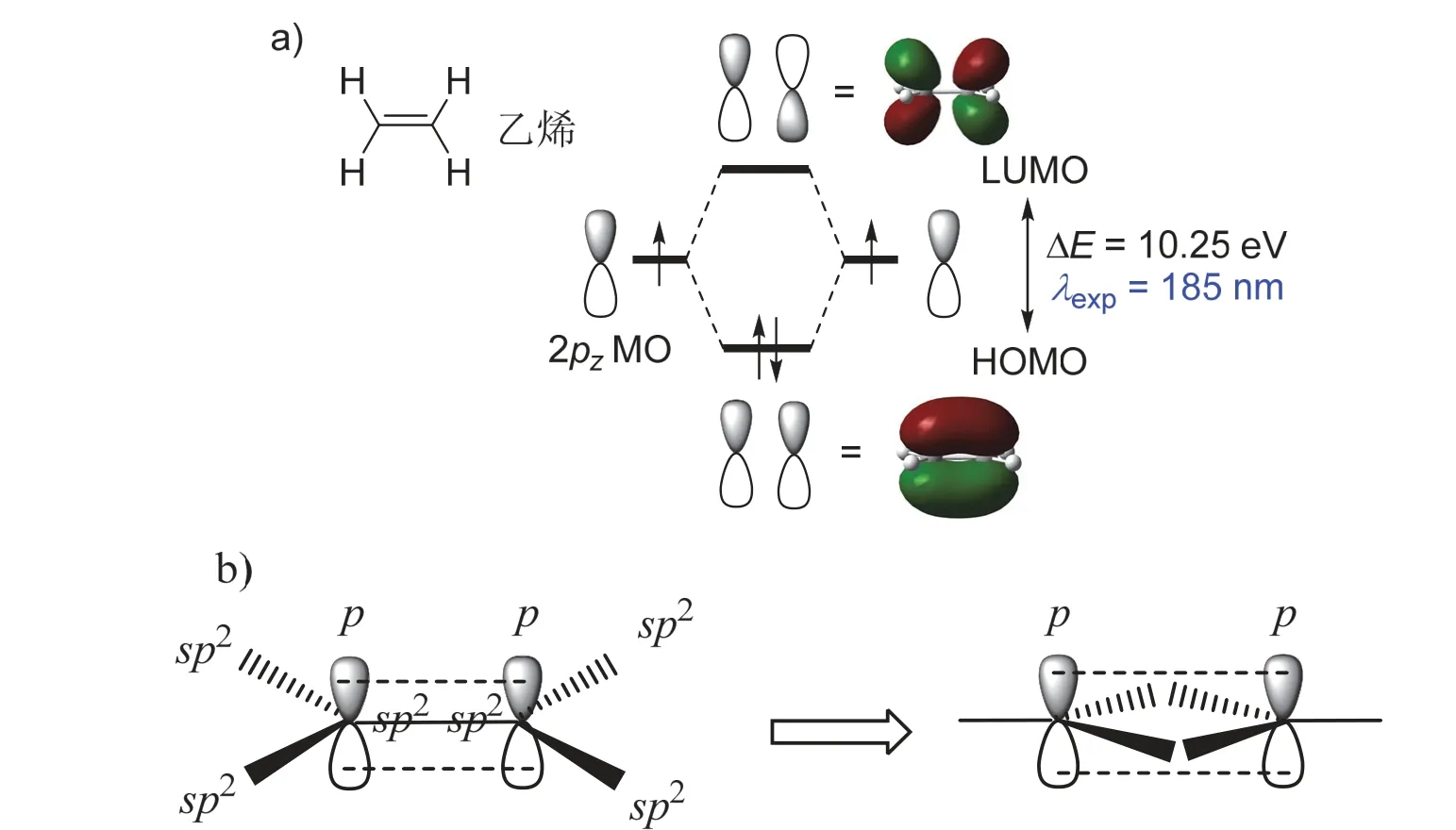
图1 a) 乙烯分子p轨道形成π成键(HOMO)和π反键轨道(LUMO)的示意图(isovalue = 0.06); b) 杂化轨道分别与其他原子或原子团成σ键,而C、C之间只有两个p轨道交叠,形成独立的π单键
2 独立存在的C―π―C单键
2017年,日本广岛大学的著名化学家Manabu Abe就发表了一篇名为“Isπ-Single Bonding (C―π―C) Possible? A Challenge in Organic Chemistry”的综述类文章[1]。文中提到没有σ键的支撑而单独存在的π键是可能的。相比正常的π键,这类单独存在的π单键一般键能更弱,反应活性更高,因此很难被分离和表征。
Abe教授课题组一直致力于分离单独存在的含碳π单键的新物种,近年来他们课题组终于发现了一类低温下可稳定存在的物种,例如化合物1 (图2)[2-6]。由于反应活性极高,室温时该化合物的寿命仅为320 ns。在M062X/def2TZVP等级上的密度泛函理论计算(Density Functional Calculations,DFT)[7]表明该化合物的HOMO主要由C1和C3两个间位碳原子上的p轨道电子重叠组成,相应的LUMO也集中在这两个碳原子上,并且与上述乙烯中的π成键和π反键轨道非常相似。通过自然键轨道分析(Natural Bond Orbital,NBO)可知[8],这两个碳原子之间的韦伯格键级(Wiberg bond index,WBI)为0.31,这和文献在(2/2) CASSCF/6-31G(d)等级上的计算结果(0.37)吻合。以上理论计算结果指出化合物1上的C1和C3之间的π电子共轭程度不高,远低于乙烯分子碳碳双键的WBI值2.05,即乙烯中π键键指数接近1。这也和相应分子的前线轨道能级差ΔE(10.25 eV (乙烯) > 2.99 eV (1))和紫外-可见吸收光谱(Ultraviolet-Visible Absorption Spectrometry,UV-Vis)中的最大吸收波长的实验数据λexp(185 nm (乙烯) < 571 nm (1))对比结果吻合。综上所述,虽然化合物1有着部分未成键的双自由基特性(1a,图2),但0.31的键级亦表明了该化合物含有独立存在的C―π―C单键(1b,图2)。
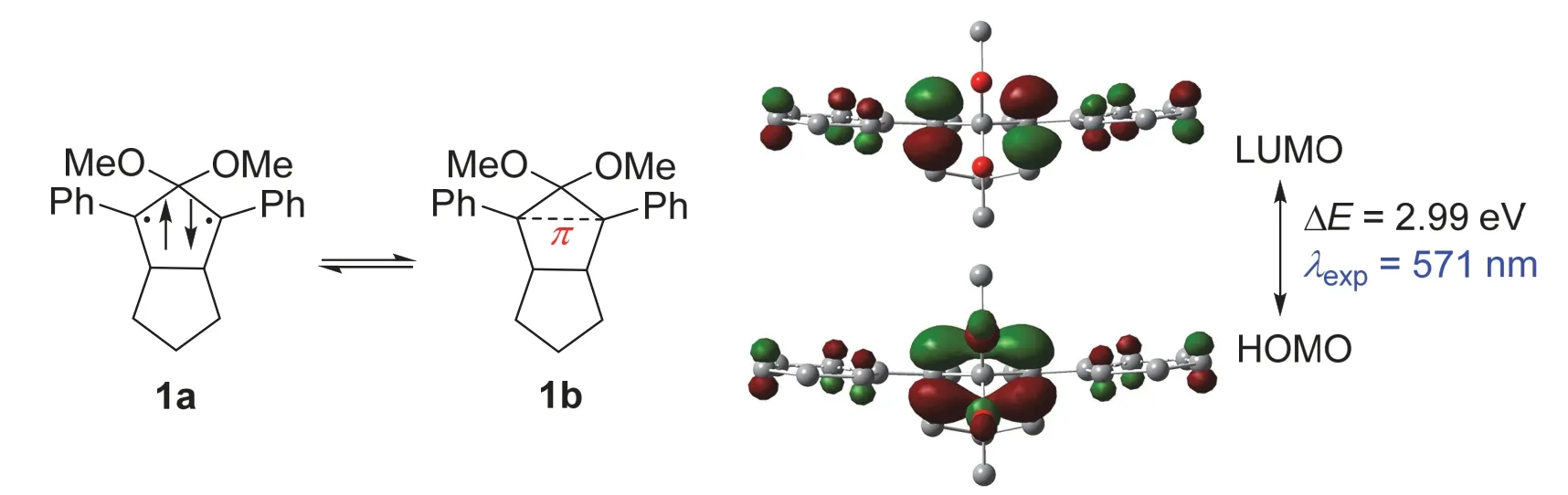
图2 单独存在的含C―π―C单键的有机物种1,及其HOMO和LUMO轨道示意图(isovalue = 0.06)
3 独立存在的B―π―B单键
由于反应活性过高,室温下能稳定存在的基于碳元素的含独立π单键的有机物还未见诸报道。幸运的是,精心设计合成的基于其他主族元素的含独立π单键的有机物表现出良好的稳定性,其不但于室温下稳定,而且可以通过单晶衍射对其结构进行精确表征。
2002年,美国加利福尼亚大学的Guy Bertrand教授报道了一种室温稳定的B2P2四元杂环双自由基(2a,图3)[9]。单晶衍射表明该化合物的四元环处于一个完美的平面结构,并且每个P―B键键长都几乎相等(0.189 nm)。当用位阻比较小的取代基时,两个硼自由基之间将会键合而生成[1.1.0]双环结构杂环,所以大位阻取代基提供的动力学稳定性对这个化合物而言非常重要[10]。最让人惊讶的是化合物2中对角的B―B键键长非常长(0.257 nm),比文献报道的最长B―B单键(0.186 nm)还长38%。这些结果指出两个B原子之间可能不存在相互作用,这样化合物2就是一个双自由基。然而,DFT计算表明两个B原子之间有着比较强的相互作用(图3)。化合物2的HOMO是主要由两个B原子的p轨道的交叠组成的π轨道,而其LUMO也集中在这两个B原子上,并且其形状类似于一个π反键轨道。进一步的NBO分析指出两个B原子之间的WBI为0.39,略高于化合物1的WBI (0.31)。DFT计算结果认为化合物2是一个存在独立含硼π单键的新型物种2b。因此,化合物2与化合物1有着类似的情况,既含有部分的双自由基特征,又有着成独立的B―π―B单键的成分,且键级高于1中的计算值[11]。
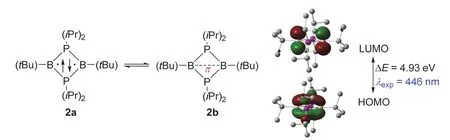
图3 单独存在的含B―π―B单键的有机物种2,及其HOMO和LUMO轨道示意图(isovalue = 0.06)
4 独立存在的Si―π―Si单键
2020年,日本东北大学的Takeaki Iwamoto教授课题组合成了一例基于四硅双环的含独立π单键化合物3 (图4)[12]。单晶衍射表明该化合物的双环结构中的桥连的Si―Si键键长为0.25822 nm,这明显比正常Si―Si共价单键(0.232 nm)[13]要长,但是比目前报道最长的Si―Si共价单键(0.2697 nm)[14]要短。这两个桥连的硅原子呈现出完全平面的几何构型,即环绕硅原子的键角和为360.00°。碘原子有少许偏离Si4环,∠ISiSi键角为178.49°。两个五元杂环中硅原子之间的距离为0.3903 nm。单晶结果分析指出这个化合物中可能含有两个硅自由基,或者存在Si―Si弱相互作用。化合物3的核磁谱图具有较好的分辨率,表明这个化合物中电子自旋态是单线态。进一步的DFT计算则证实了其基态为单线态。
化合物3的前线轨道如图4所示,其中HOMO主要离域在两个桥连的硅原子上,并且是由两个硅原子的p轨道的交叠组成的π轨道,而LUMO主要是集中在这两个硅原子上的典型π反键轨道。NBO分析表明这两个硅原子之间的WBI为0.80,和文献报道的0.77非常接近[12]。DFT计算结果表明这两个硅原子之间存在显著的Si―π―Si单键共价相互作用。
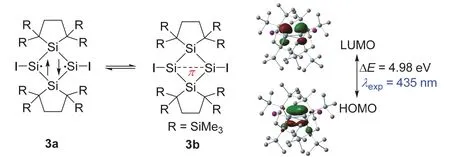
图4 单独存在的含Si―π―Si单键的有机物种3,及其HOMO和LUMO示意图(isovalue = 0.06)
同年,日本群马大学(Gunma University)的Hideyuki Matsumoto教授等人也发现了一例新的含有单独存在的含Si―π―Si单键的有机物种,其中Si―π―Si键键长为0.2853 nm[15]。通过X射线晶体学、电子顺磁共振、磁化率、UV/Vis和29Si NMR光谱实验,以及包括自然键轨道分析在内的理论计算,该团队证明桥头Si原子之间存在Si―π―Si单键。然而,在2021年6月份,波兰科学院有机化学研究所的Cina Foroutan-Nejad助理教授从拓扑学的角度分析了这个问题,并提出了不一样的观点[16]。他指出,经典分子轨道(Canonical Molecular Orbitals,CMOs)分析可以证实σ键的存在,其中HOMO-6代表两个sp2杂化的Si原子之间的σ相互作用。此外,电子密度的拓扑及定量分析表明,Si原子之间同时存在π键和相对较弱的但不可忽略的σ键。因此,Foroutan-Nejad认为,Matsumoto教授提出的Si―π―Si单键应为一个独特的π键比σ键更强的双键系统。对于Foroutan-Nejad的质疑,Matsumoto等人则表示期待更多类似的探讨。在科学的道路上,这样的探讨并不少见,也正是这些思想的碰撞,才使科学的进步具有源源不断的动力。
5 结语
从上述已经报道的例子中,我们可以总结出含独立存在π单键的化合物,其最大吸收波长都明显长于含经典π键的化合物,如571 nm (1) > 446 nm (2) > 435 nm (3) > 185 nm (乙烯),这也和计算得到的LUMO和HOMO能量差值次序相吻合(图1-4)。相应的,所有独立存在的π单键,其键长都显著长于其理论上的共价单键,表明其键能比经典π键要低。这也是为什么含独立存在π单键的化合物稳定性差,难以分离,迄今为止只有3例含部分独立存在π单键的稳定化合物得到报道。此外,独立存在的π单键一般还具有以下特征:1) 都存在于平面的环状刚性结构中;2) 需要大位阻取代基提供一定程度的动力学稳定性;3) 间隔独立存在π键的原子一般都是sp3杂化中心。因此,通过对独立π单键的介绍和分析既让学生和教师更好地理解了分子价键与分子轨道理论,还为相关科研工作者提供了设计合成含独立π单键化合物所需要关注的成键特征。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
井冈山大学学报(自然科学版)(2022年2期)2022-03-31
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
水泵技术(2021年4期)2021-01-22
石油化工技术与经济(2021年4期)2021-01-13
读写算·教研版(2016年8期)2016-05-07
中学化学(2014年4期)2014-09-09
自动化博览(2014年9期)2014-02-28
