
Involvement of Met receptor pathway in aggressive behavior of colorectal cancer cells induced by parathyroid hormone-related peptide
2022-07-30 10:02NovoaazMBCarriereGigolaZwengerAOCalvoGentili
World Journal of Gastroenterology 2022年26期
Novoa Díaz MB, Carriere P, Gigola G, Zwenger AO, Calvo N, Gentili C
Abstract
Key Words: PTHrP; MET receptor tyrosine kinase; Parathyroid hormone receptor type 1; Colorectal cancer;Drug resistance
lNTRODUCTlON
Colorectal cancer (CRC) is one of the most frequent tumors of the digestive system and represents the third type of cancer with the highest incidence worldwide[1].According to statistics, the development of metastasis and resistance to chemotherapeutic drugs causes 90% of deaths in patients with CRC[2].Although treatment strategies have been improved in the past years, the mortality associated with this oncological disease is still around 50%[1,3]. In this line, the World Cancer Research Fund International indicates that CRC incidence is still increasing and that its prevalence is expected to augment by 60% in the next few years[4]. This is mainly due to the emergence of CRC patients under 50 years old and the appearance of new tumors subtypes that have a refractory response to the usual therapy[5]. Currently,two of the chemotherapeutic agents approved as first and second-line palliative drugs in CRC are oxaliplatin (OXA) and irinotecan (CPT-11)[6,7]. However, more than half of patients with stage II/III disease who underwent surgery and were treated with these drugs relapsed and died[8]. Thus, all these facts evidence an urgent need to elucidate the molecular mechanisms associated with two key aspects of therapy: Effectiveness and resistance.
Parathyroid hormone-related peptide (PTHrP) is a cytokine-like protein that is normally produced in many adult and fetal tissues[9,10]. The N-terminal 13 amino acid residues of PTHrP exhibit high structural homology with the PTH[11]. This allows the binding of both ligands to the same G-proteincoupled receptor, PTH/PTHrP type 1 (PTHR1)[12,13].It is known that PTHrP and its receptor are expressed in the same cells or neighboring cells. This juxtaposition is directly related to its activity as an autocrine and paracrine factor[14,15].
Regarding the expression of this protein in neoplastic disease, studies in patients show its overexpression in tissues of breast, prostate, lung, and colon cancers[16,17]. Moreover, 95% of colorectal adenocarcinomas have elevated expression of this protein[18-20]. Although PTHrP was initially identified as a major contributor to hypercalcemia in paraneoplastic syndromes, it is known that this peptide also plays a key role in the development and progression of many tumors [10,16,21-23]. In this concern, in CRC-derived cells we found that PTHrP stimulated cell proliferation, survival, and migration, the epithelial-mesenchymal transition (EMT), the release of pro-angiogenic factors, and the resistance to CPT-11[13,24-26]. In xenografts of CRC cells, we have observed that PTHrP also modulates the expression of molecular markers linked to tumor progression[24-27]. Despite these contributions,relevant aspects from the action of this peptide are still unknown, specifically, whether PTHrP could promote resistance to other forms of chemotherapy and the validation of our previous observations using human samples.
Met is a receptor with tyrosine kinase (RTK) activity, and it is expressed in normal tissues and participates in various physiological processes such as embryonic development and wound repair[28].Only hepatocyte growth factor (HGF) is known as a necessary ligand for its activation. However, crosscommunications between this RTK and G-protein-coupled receptors were reported to promote its aberrant activation[29-31]. Met overexpression or its dysregulation can lead to malignant cell transformation and contributes to the development and progression of different types of cancer including colorectal tumors[28,32-35]. Moreover, Met dysregulation is also associated with drug resistance in colon cancer cells[32,35,36]. Several studies demonstrated the overexpression of this RTK in tumor tissue of CRC patients; in this regard, its inhibition is being widely investigated as a complementary treatment to usual therapies[37,38]. Despite this, the value of Met as a prognostic marker or in treatment strategies has not been established yet.
Although empirical evidence suggests that Met and PTHrP play a key role in the development and evolution of CRC, it is still unknown whether PTHrP and Met act cooperatively to promote events associated with the progression and chemoresistance in CRC. Based on our previous studies, we hypothesized that the signaling pathway trigged by PTHR1 after binding to PTHrP could be involved in the transactivation of the RTK Met and consequently in the aggressive behavior of CRC cells. For this reason, the present work aimed to elucidate the relationship among PTHR1, PTHrP, and Met.
MATERlALS AND METHODS
Reagents and antibodies
Human PTHrP (1-34) and trypan blue dye were obtained from Sigma-Aldrich Chemical Co (St. Louis,MO, United States). Antibodies were purchased from the following sources: Anti-Met and anti-phospho Met (Tyr 1234/1235) and anti-E-cadherin were provided by Cell Signaling Technology (Beverly, MA,United States); anti-PTH/PTHrP receptor type 1, anti-GAPDH, goat anti-rabbit peroxidase conjugated secondary antibody, and goat anti-mouse peroxidase conjugated secondary antibody were from Santa Cruz Biotechnology (Santa Cruz, CA, United States). PD98059, SB203580, and PP1 were obtained from Calbiochem (San Diego, CA, United States) and SU11274 from Sigma-Aldrich Chemical Co. RNase CocktailTM Enzime Mix was from Applied Biosystems (Carlsbad, CA, United States). Protein size markers were from Amersham Biosciences (Piscataway, NJ, United States). Immobilonpolyvinylidene difluoride (PVDF) membranes and electrochemiluminescence (ECL) detection kit were from Amersham(Little Chalfont, Buckinghamshire, England). The chemotherapeutic drugs CPT-11, OXA, and doxorubicin (DOXO) were generously provided by Dr. Ariel Zwenger.
Cell culture and treatment
Human colon cancer cells Caco-2 and HCT116 (American Type Culture Collection, Manassas, VA,United States) were grown at 37 °C in an atmosphere containing 5.5% CO2in Dulbecco’s Modified Eagle Culture Medium (DMEM) containing 10% heat-inactivated and irradiated fetal bovine serum (FBS), 1%non-essential amino acids, penicillin (100 IU/mL), streptomycin (100 mg/mL), and gentamicin (50 mg/mL). The cells were cultured until reaching 80% confluence and then they were deprived of FBS for 2 h. Treatments were performed by incubating cells with 2.5% FBS-DMEM in the absence or presence of PTHrP (1-34) (10-8M) at different times. This dose was selected based on previous studies[39,40]. Where indicated, cells were pre-incubated for 30 min with PD-98059 (20 μM), an inhibitor of the mitogenactivated protein kinase (MAPK) kinases (MEKs), the upstream kinases of ERK1/2; SB203580 (20 μM),an inhibitor of p38 MAPK; PP1 (10 μM), an inhibitor of Src; or SU11274 (0.5 μM), a Met inhibitor. In previous works, we confirmed the effectiveness of the inhibitors PD98059, SB203580, and PP1[26,27],whereas SU11274 dose was chosen in accordance with the literature[41]. Controls were run by the addition of an equivalent volume of dimethylsulfoxide (DMSO), the vehicle of the inhibitors.
Western blot analysis
Cells were washed with phosphate buffered saline (PBS) with 25 mmol/L NaF and 1 mmol/L Na3VO4and lysed in buffer containing 50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 3 mmol/L KCl, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1% Tween 20, 1% Nonidet P-40, 20 μg/mL aprotinin,20 μg/mL leupeptin, 1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 25 mmol/L NaF, and 1 mmol/L Na3VO4. Lysates were vortexed for 45 s, incubated on ice for 10 min, and then centrifuged at 14000gand 4 °C for 15 min. The supernatant was collected and protein quantification was performed by the Bradford method[42]. The proteins present in the samples were separated (30 μg/lane) using sodium dodecyl sulfate (SDS)-polyacrylamide gels (8%-10% acrylamide) and electrotransferred to hydrophilic polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% skim milk in Tris buffered saline-Tween (TBS-T) buffer (50 mmol/L Tris, pH 7.2-7.4, 200 mmol/L NaCl, 0.1%Tween-20), and then incubated overnight with the appropriate dilution of primary antibody (Met #8198 1:3000, p-Met #3077 1:500, E-cadherin #3195 1:1000; GAPDH sc-32233 1:5000) in TBS-T with 2.5% bovine serum albumin. After washing, the membranes were incubated with the appropriate dilution of horseradish peroxidase conjugated secondary antibody in TBS-T with 2.5% skim milk. Finally, proteins were revealed using a commercial electrochemiluminescence kit, and the bands obtained were digitized densitometrically and quantified with Image J program.
Stripping and reprobing membranes
To remove primary and secondary antibodies, membranes were incubated in stripping buffer (62.5 mmol/LTris-HCl pH 6.8, 2% SDS, and 50 mmol/L β-mercaptoethanol) at 55 °C for 30 min in agitation,washed for 10 min in TBS-T (1% Tween-20), and then blocked, as indicated above. After this procedure,it is possible to re-test antibodies in the membranes.
RNA isolation and cDNA synthesis
HCT116 cells were incubated in serum-free DMEM for 2 h and then treated with or without PTHrP (10-8mol/L, in DMEM 2.5% FBS) for 15 min. Total RNA from all samples was isolated using the High Pure RNA Isolation Kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions. Total RNA (1 g) from each sample was used for first-strand cDNA synthesis using High Capacity cDNA Reverse Transcription Kits following the manufacturer’s instructions (Applied Biosystems, United States). The cDNA product was quantified and then stored at -20 °C for real-time polymerase chain reaction (RT-qPCR).
RT-qPCR
PCR reactions were carried out on a real-time PCR system (Applied Biosystems, model 7500). The primers used are the following: 5’-GGAAACACCCATCCAGAATGTCATT-3’ (forward) and 5’-TGATATCGAATGCAATGGATGATCT-3’ (reverse) forMet; 5’-ACCACAGTCCATGCCATCA-3’(forward) and 5’-TCCACCACCCTGTTGCTGTA-3’ (reverse) forGAPDH. The PCR reactions were prepared using SYBR green master mix (No. 4309159, Applied Biosystems). On ice, the following was added: 5 μL of 2 × SYBR green, 0.8 μL of forward primer (400 nmol/L), 0.8 μL of reverse primer (400 nmol/L), 2 μL of template cDNA (12 ng), and 1.4 μL of Milli Q sterile water.GAPDHcDNA was amplified in parallel for all genes to provide an appropriate internal PCR control. For each experiment,water was added as a sample without a template (negative control). Mean Ct of the gene of interest(Met) was calculated from triplicate measurements using the following equation: Ct =Ct of the gene of interest -Ct ofGAPDH. mRNA levels were calculated by the 2-ΔCTmethod.
Wound healing assay and morphological monitoring
Once confluence was reached, a cross-shaped wound was made manually in the cell monolayer with a tip and two washes were performed with sterile 1 × PBS to remove debris from the procedure. Then, the treatments were performed in DMEM without FBS as detailed above. Finally, the wounds were visualized under an inverted microscope and each of them was photographed at 0 h, 5 h, and 24 h,where the same fields were photographed. For each condition, the closure of the wound concerning the control was analyzed using ImageJ-NIH program, which allows for quantifying the area that remains cell-free.
To evaluate morphological changes potentially associated with the EMT, cells were treated with PTHrP as detailed above and the same fields were observed with a microscope and photographed during the times when significant molecular changes were observed.
Measurements of drug effects of chemotherapeutics
CPT-11, OXA, and DOXO cytotoxicities were assessed by trypan blue dye exclusion test. HCT116 cells were seeded in a 24-well plate until 80% confluence and then treated with or without PTHrP 10-8M and/or CPT-11(10 μM)/OXA (10 μM)/DOXO (5 μM) in triplicate for 24 h. The doses of these drugs were chosen according to previous works[26,43-45].Where indicated, HCT116 cells were pre-incubated with SU11274 (0.5 μM)[41] or DMSO, the vehicle of the inhibitor. After the treatment, cells were washed with PBS and then incubated with trypsin-EDTA to separate them from the culture substrate. The cell suspension was diluted in equal volume with trypan blue stain (1:1). The number of cells per field was counted in a Neubauer chamber; cells that exclude the dye were considered viable cells. The number of viable cells was calculated according to the following formula: Total number of viable cells = average number of viable cells × dilution factor × 104[46].
Xenograft in nude mice
Xenografts on 6-wk old male N:NIH (S)_nu mice (body weight 20-25 g) were generated through a subcutaneous injection of 1 × 106HCT116 cells in their left dorsal flanks[47]. Four days after this procedure, the animals were randomly separated into treated and control groups (n= 6/group). Mice received a daily intratumoral injection of PTHrP (40 μg/kg) in 100 μL PBS, or PBS as a control. This manner of administration was in order to keep the compound level constant in the tumor area. The dose of PTHrP was selected according to previous studies performed by us and by other investigators with PTH and/or PTHrP in rodents[27,48-50]. Throughout the experiment, animals were kept under strict sanitary barriers in insulated cabins and had access to sterilized food and waterad libitum. Besides, they were exposed to 12 h cycles of light and darkness (200 Lux/1 m from the floor), atmospheric pressure,25 °C, and humidity of 40%-60%. Humanitarian slaughter was carried out after 20 d of treatment by inhalation of the chemical agent CO2. The tumors were removed, weighed, and fixed in a 4% formaldehyde solution and included in a paraffin block for subsequent treatment in immunoassays. At the end of the trial, the average weights were 0.30 g ± 0.11 g (standard deviation) and 0.40 g ± 0.10 g for control tumors and treated tumors, respectively. Whereas the average volumes were 0.21 mm3± 0.08 mm3(standard deviation) and 0.26 mm3± 0.08 mm3for control tumors and treated tumors, respectively. The present animal protocol was approved by the Institutional Committee for the Care and Use of Experimental Animals from the Universidad Nacional del Sur (CICUAE-UNS, institutional endorsement updated to 2021) and it was executed in accordance with the National Institutes of Health guide for the care and use of laboratory animals (NIH 1996) in order to minimize pain or discomfort for the animals.
Patients and clinical specimens
The following research protocol (File number 8610-017183/2018, Registry of Investigations in Health(RIS) number N° 11.00.18) has the endorsement of the Advisory Commission on Biomedical Research in Beings Human Rights (CAIBSH) and the approval of the Health Ministry of the Province of Neuquén,Argentina (provision 0088-18-1-19) and was authorized by the Ethical Committee of the Hospital Interzonal de Graves y Agudos Dr. José Penna, Buenos Aires, Argentina in accordance with article 23 of Law 11044 of the Ministry of Health and the Central Research Ethics Committee (CECI) on research with biological samples from Argentina.
We performed an analysis of specimens obtained from patients with colorectal adenocarcinoma which were admitted to the Hospital Interzonal de Graves y Agudos Dr. José Penna (Bahía Blanca,Buenos Aires, Argentina) and the Hospital Provincial de Neuquén (Neuquén, Neuquén, Argentina)from January 1990 to December 2007. Normal colorectal tissue samples were assigned to a control group. The medical record of each patient was analyzed for the information as follows: Sex; age;primary tumor location; histological grade; and initial stage (according to the TNM staging system issued by the American Joint Committee on Cancer-AJCC and International Union for Cancer Control-UICC). The exclusion criteria were: Patients under 18 years of age due to the low prevalence of this cancer in the child and youth population[51]; pregnant patients due to the fact that high levels of PTHrP have been observed in the placenta and fetal tissues[52]; lactating patients because high levels of PTHrP have been detected in breast tissue and in breast milk and correlate with serum PTHrP levels in these women[53]; patients who have previously developed other types of tumors (liver, lung, and bone) since CRC commonly metastasizes to these sites. These considerations are necessary to exclude cases where the cytokine derived from other tissues may interfere with the analysis of the results from this study. All are exclusion criteria at the time of diagnosis. The confidentiality criteria were based on the regulatory provisions referring to confidentiality in clinical investigations, including Law 11044 and Law 25326 on data protection, from Argentina, the Guide for Health Research of the National Ministry of Health, the UNESCO Declaration on the Protection of Genetic Data and the Declaration of Helsinki of the World Medical Association (version 2008), and the Council for International Organizations of Medical Sciences(CIOMS) Guidelines. This study respected the dignity and rights of the patients. All data derived from the clinical history was treated under strict confidentiality to protect and preserve the patient’s identities.
Paraffin-embedded archival blocks from primary tumors were reviewed by hematoxylin-eosin staining technique; subsequently, representative histological samples of each patient were selected for immunoassays.
Immunohistochemistry
From each paraffin block, tumor sections were sliced and prepared for immunohistochemical detection using the primary monoclonal antibody anti-Met (#8198, 1:2000) or anti-PTHR1 antibody (sc-12722-3D1.1, 1:50) according to the manufacturer’s protocol. The sections were deparaffinized with xylol for 15 min and subsequently rehydrated in decreasing concentrations of alcohol. Antigenic retrieval was carried out using a pressure cooker with sodium citrate buffer (10 mmol/L, pH 6) for 15 min at 1 atm.The sections were washed twice with PBS and we proceeded to block endogenous peroxidase with 30%H2O2for 10 min. After two washes with PBS, the corresponding antibody was added to each sample and incubated overnight at 4 °C. Immunohistochemical staining was carried out manually using ABCAM Detection IHC Kit (Abcam, Cambridge, MA, United States) according to the manufacturer’s instructions. The images obtained under an optical microscope were analyzed using an open-source image processing package based on ImageJ-NIH program[54,55].
Statistical analysis
To obtain statistical data, three experiments were carried out independently and the results are expressed as the mean ± SD. To determine significant differences between two groups of data, Student’st-test (two-tailed, equal variance) was carried out. APvalue < 0.05 was considered statistically significant.
For the statistical analysis of human tumor samples, the correlation coefficient between the expression of Met and PTHR1 was evaluated by the Spearman test. Then, the patients were grouped according to clinicopathological characteristics such as age, sex, TNM stage (II, III, and IV), location of the primary tumor (right colon, left colon, and rectum), and histological grade (G1, G2, and G3). The Fisher’s exact test was used to evaluate the association of Met or PTHR1 expression with each variable. These statistical analyses were performed using the R programming language version 4.1.1[56]. APvalue <0.05 was considered statistically significant.
RESULTS
PTHrP modulates the expression of the RTK Met in human HCT116 cells
Met is a heterodimeric receptor that is composed of two chains, alpha and beta. Initially, this RTK is synthesized as a single-chain precursor (pro-Met), and it then undergoes posttranslational modifications to produce the mature form[57]. The binding of the HGF to the beta subunit of Met induces its dimerization and the phosphorylation of tyrosine domains in the intracellular portion of the receptor(Y1234 and Y1235). These events promote the kinase activity of Met and its autophosphorylation (Y1349 and Y1356). Then, various proteins are recruited to these phosphorylated sites, triggering different signaling pathways[33,58].
In CRC cells, Met signaling is activated by various mechanisms to promote tumor development and progression[28,59]. Gene amplification or mutations (that lead to receptor overexpression) and increased activity due to ligand-mediated stimulation induce the aberrant activation of Met[28]. As mentioned earlier, another mechanism that favors its abnormal activation involves the phosphorylation in Met activator domains and is indirectly due to the previous activation of cell-surface receptors like Gprotein-coupled receptors[29,31].
In the past years, several authors have reported a correlation between the overexpression of PTHrP and tumor progression in lung, breast, and prostate cancers[16,21,60]. Our research group has shown that this cytokine also promotes the aggressive phenotype of intestinal tumor cells[13,24-26].This protumor factor exerts its action on CRC cells through several mitogenic signaling pathways[13]. In view of this background, in this work we decided to investigate whether the molecular mechanisms trigged by PTHR1 after binding of PTHrP are capable of modulating the expression of the mature form of the RTK Met and/or its activation in CRC-derived cells. To that end, we initially treated Caco-2 cells and HCT116 cells with PTHrP for different times and then the cell lysates were immunoblotted with an anti-Met antibody. We selected these cell lines from CRC with different aggressive phenotypes because both express the receptor PTHR1 and we had previously verified that their cell responses are mediated exclusively by this receptor[26,40,61]. Met did not change its protein expression when Caco-2 cells were treated with PTHrP over a wide interval of exposure times (data not shown). However, the response of the HCT116 cell line, which is more aggressive than Caco-2 cell line, was different. As seen in Figure 1(top and middle panels), PTHrP treatment for 30 min increased Met protein levels. However, the protein expression diminished at 3 min and 10 min, and at 1 h to 3 h of exposure to the peptide. Figure 1(lower panel) shows that at 5 h of treatment, Met protein levels continued to decrease. However, PTHrP increased Met protein expression at 20 h of treatment.
As it happens with other RTKs on the cell surface, after its phosphorylation/activation, Met is internalized with the subsequent decrease in its protein levels due to the proteasomal degradation[62,63]. In this regard, the reduction of Met protein levels observed in HCT116 cells suggested a previous activation of this receptor by PTHrP, and the increment of its protein level after these decreases (at 30 min and 20 h) suggested a modulation of Met expression by PTHrP in order to improve Met levels.
PTHrP promotes Met phosphorylation and activation through Src kinase in human HCT116 cells
Our next goal was to elucidate if the decrease in Met protein levels observed in HCT116 cell line and mediated by exogenous PTHrP could be due to its previous phosphorylation/activation. Thus, we proceeded to evaluate the status of phosphorylated Met under PTHrP action. As shown in Figure 2A,Western blot analysis using a specific antibody that recognizes active Met, revealed that PTHrP treatment for 3 min and 10 min increased the phosphorylation of Met at the residues Tyr 1234 and Tyr 1235. These phosphorylation sites constitute activating domains of the receptor. The decrease in total Met protein levels (showed in Figure 1 and pointed in Figure 2A with arrows) ruled out the possibility that an increase in phosphorylated Met levels at the same time was due to the up-regulation of Met protein expression.
Src is a cytosolic tyrosine kinase deregulated in 80% of CRC patients[64-66]. Previously, we reported the activation of Src in the HCT116 cell line induced by PTHrP at 3 min of treatment[26]. As we mentioned above and taking into account that we observed phosphorylation of Met by PTHrP on specific tyrosine residues that are needed for its activation, we decided to evaluate whether Src is involved in this process induced by the cytokine PTHrP in intestinal tumor cells. For this purpose, the cells were pre-treated with PP1, a specific inhibitor of Src activation, and then with PTHrP for 3 min. As shown in Figure 2B, we observed that PP1 decreased PTHrP-induced phosphorylation of Met at Tyr1234/1235. These findings suggested that in the HCT116 cell line, PTHrP rapidly activate the Src signaling pathway, leading to the phosphorylation and activation of Met.
PTHrP promotes the phosphorylation/activation of Met through the MAPK signaling pathway in human HCT116 cells
It has been described that MAPK signaling pathway activation is very common in CRC and is related to an aggressive phenotype of intestinal tumor cells[67]. Due to its relevance in cancer, in recent decades the possibility of employing MAPK pathway inhibitors has been studied for the treatment of this disease[68,69]. In our laboratory, we found that PTHrP induces the activation of the ERK 1/2 MAPK and p38 MAPK and that the activity of Src converges in the phosphorylation/activation of MAPK in CRC-derived cells [26,27].
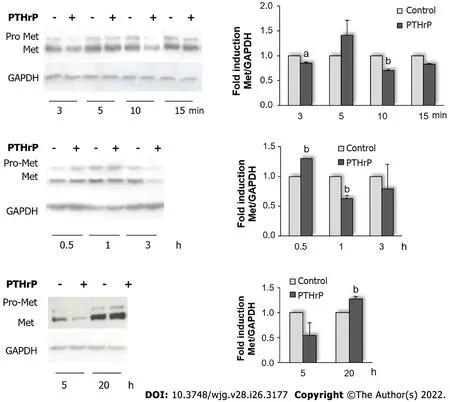
Figure 1 Parathyroid hormone-related peptide modulates the protein expression of Met receptor in HCT116 cells. Cells were treated with or without 10-8 mol/L parathyroid hormone-related peptide (PTHrP) for different times. The protein levels of pro-Met (Met precursor) and Met [tyrosine kinase receptor(RTK) mature form] were analyzed by Western blot to investigate whether the molecular mechanisms trigged by PTHR type 1 (PTHR1) after binding of PTHrP are capable of modulating the expression of the mature form of the RTK Met in HCT116 cells. GAPDH protein levels were determined as a control of the amount of proteins present in the membrane, since this protein is not substantially modified with the treatment by the cytokine. Graph bars represent the average of the results obtained from three independent experiments. GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; Met: Receptor tyrosine kinase Met; PTHrP: Parathyroid hormone-related peptide. aP < 0.05; bP < 0.01.
Based on our previous results, we next decided to investigate in our model if ERK 1/2 MAPK and p38 MAPK are also involved in the effects of PTHrP on the Met signaling pathway. To that end, the cells were pre-incubated with PD-98059 (a selective inhibitor of MEK, the upstream kinases of ERK 1/2) or with SB-203580 (a specific p38 MAPK inhibitor) and then exposed to PTHrP for 1 h. This time point was selected because the phosphorylation/activation of ERK 1/2 MAPK and p38 MAPK significantly increased at 60 min after PTHrP treatment in HCT116 cells[26,27]. Regarding the cell response when tumor cells were exposed to PTHrP for 1 h (Figure 1), Figure 3 shows an increase in the phosphorylated Met levels, suggesting that the treatment with the cytokine for 1 h also induces the phosphorylation/activation of Met with the subsequent reduction in its protein levels due to its proteasomal degradation.Moreover, Figure 3 reveals that the selective inhibition of MEK MAPK and p38 MAPK averted the effect on Met phosphorylation, suggesting that Met activation is regulated by the ERK 1/2 MAPK and p38 MAPK signaling pathways in HCT116 cells under PTHrP action.
PTHrP increases Met gene expression in human HCT116 cells
As it was commented earlier, the fact that Met, like other RTKs, undergoes proteasomal degradation after its phosphorylation/activation[62,63] explains that at the same time points of PTHrP exposure (3 min, 10 min, and 60 min), the levels of phosphorylated Met increased while the protein levels of total Met decreased. Furthermore, in this work we elucidated that Met is phosphorylated by PTHrP through the Src and MAPK signaling pathways. In view of the results shown in Figure 1, we selected a time before 30 min with the purpose to evaluate whether the increase in Met protein levels initially observed by PTHrP treatment is related to the modulation of gene expression. By real-time PCR, we analyzed theMetmRNA levels and found that PTHrP increasedMetgene expression at 15 min (Figure 4). This result suggested that in HCT116 cells, PTHrP at least at this time promotes the transcription of theMetgene and its translation, correlating with the increase of the protein levels observed at 30 min of PTHrP exposure.
Met signaling pathway induced by PTHrP participates in cell events related to the aggressive behavior of human HCT116 cells
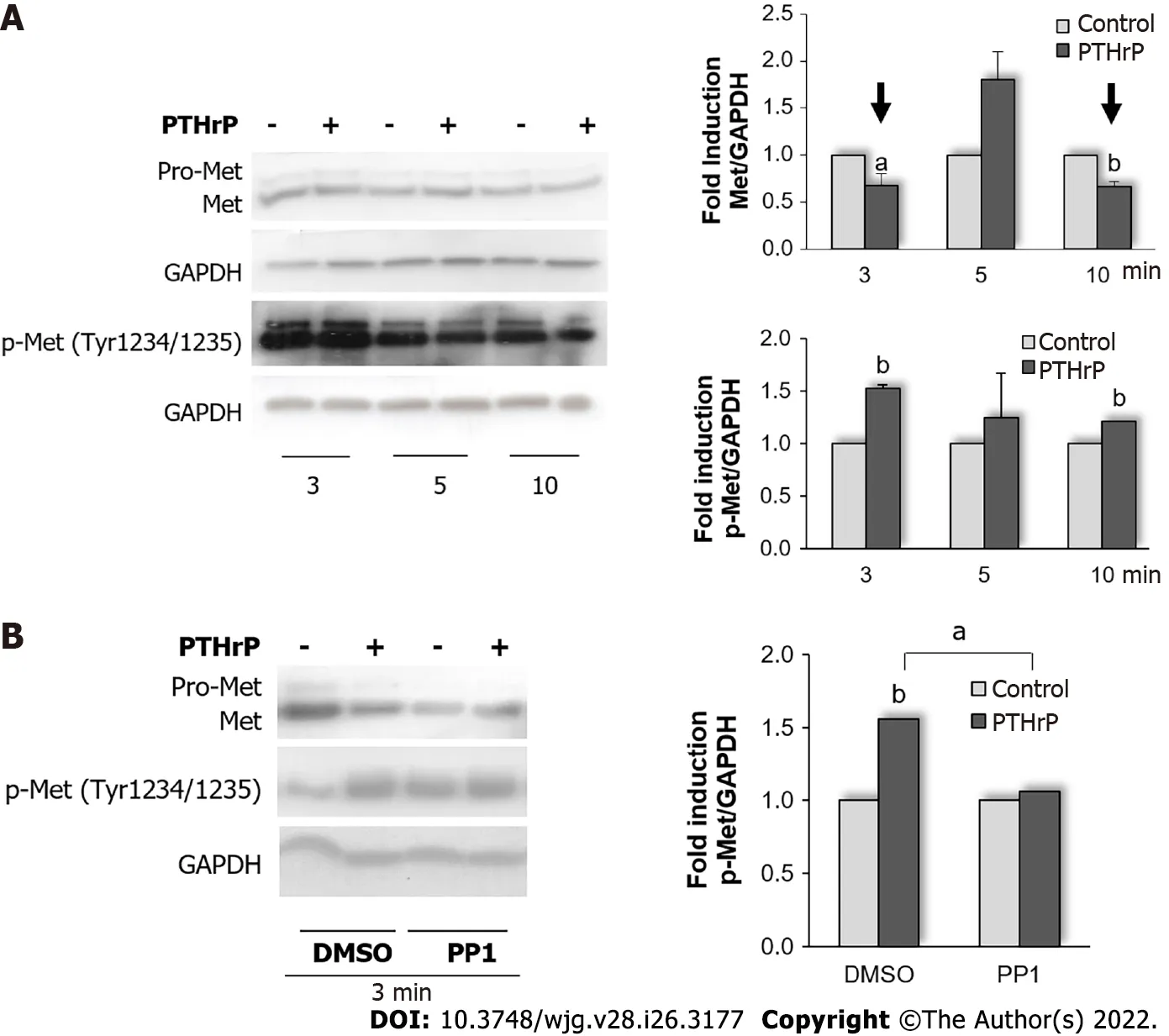
Figure 2 Parathyroid hormone-related peptide increases Met phosphorylation and activation through Src kinase in HCT116 cells. A: Cells were exposed to 10-8 mol/L parathyroid hormone-related peptide (PTHrP) 10-8 mol/L for 3 min, 5 min, and 10 min. The protein levels of Met and p-Met (phosphorylated in the residues Tyr1234 and Tyr1235) were assessed by Western blot to investigate whether PTHrP is capable of modulating Met phosphorylation and activation in HCT116 cells. Graph bars represent the average of the results obtained from three independent experiments; B: Cells were pre-treated with PP1, a selective Src inhibitor, for 30 min and then exposed to 10-8 mol/L PTHrP for 3 min to evaluate whether Src kinase mediates the effect of PTHrP on Met activation in HCT116 cells.Controls were run by adding an equivalent volume of DMSO, the vehicle of the inhibitor. The protein levels of p-Met and Met were evaluated by Western blot. Graph bars represent the average of the results obtained from three independent experiments. In all experiments, GAPDH protein levels were determined as a control of the amount of proteins present in the membrane, since this protein is not substantially modified with the treatment by the cytokine. DMSO: Dimethylsulfoxide; GAPDH:Glyceraldehyde 3-phosphate dehydrogenase; Met: Receptor tyrosine kinase Met; p-Me: Phospho-Met (Tyr1234/1235); PTHrP: Parathyroid hormone-related peptide.aP < 0.05; bP < 0.01.
Our studies in CRC cell models have demonstrated that PTHrP favors several events associated with the progression of the disease, such as cell proliferation, migration, and the EMT program[13,24,26,27,39].Based on these previous findings and since Met is modulated by PTHrP in HCT116 cells, we decided to evaluate whether the Met signaling pathway participates in events induced by the cytokine and associated with an aggressive phenotype of CRC-derived cells. For this purpose, cells were incubated with SU11274, an ATP competitive inhibitor of the catalytic activity of the Met kinase[70], followed by the treatment with or without PTHrP for different times, and then we performed different procedures as described below. The times of PTHrP exposure were selected based on our previous findings[24,26,27].As shown in Figure 5A, PTHrP or PTHrP plus DMSO (the vehicle of the inhibitor) at 24 h increased the number of HCT116 cells. However, the treatment with the Met inhibitor significantly decreased the number of viable cells concerning the control. As the Met signaling pathway is involved in the proliferation and survival of intestinal tumor cells[41,71-73], this descent in the cell viability suggests that both cell responses are affected.
By the wound healing assay, we evaluated whether Met signaling is also involved in the migration of HCT116 cells induced by PTHrP. We observed the wound healing in the culture of confluent HCT116 cells to compare the migration between untreated cells and cells treated with PTHrP in the presence or absence of SU11274. Representative photographs, taken at time points 0 h, 5 h, and 24 h of the identical location and the quantification of the results of two separate experiments are shown in Figure 5B. A significant enhancement in wound closure was detected in cells exposed to PTHrP or PTHrP plus DMSO compared to the control at 5 h and 24 h. However, this effect was not observed in cells that had been pre-treated with the Met inhibitor.
We recently published results revealing that PTHrP is able to induce the EMT in HCT116 cells[24].The EMT is a key program of CRC that participates in the invasion, angiogenesis, and chemoresistance associated with metastasis[74,75]. Based on this previous work, herein we analyzed if PTHrP also induces this program through the Met pathway. To that end, HCT116 cells were incubated with SU11274 and then treated with or without PTHrP. The cells were observed under an inverted microscope after treatment with PTHrP for 5 h and 24 h to analyze the morphological changes associated with the EMT. The arrows in Figure 5C point to morphological changes corresponding to the transition from a polygonal structure to spindle-like structure in cells treated for 24 h with PTHrP or PTHrP plus DMSO. These findings revealed an effective transition to the mesenchymal phenotype and are consistent with our previous work in HCT116 cells[24]. There were no changes between SU 11274 control cells and those treated with the inhibitor of Met plus PTHrP. To further evaluate the phenotype related to a mesenchymal shape, we measured the minor and major axes in the photomicrographs using the Image J-NIH program. Figure 6 reveal that under PTHrP action, the cells lost their epithelial characteristics. The increase of the relation between the major and minor axes indicated that the exposure to PTHrP for 24 h significantly increased the degree of HCT116 cell elongation, a typical feature of the mesenchymal shape. However, when the cells were pre-incubated with SU11274, the effects of the cytokine were reverted.
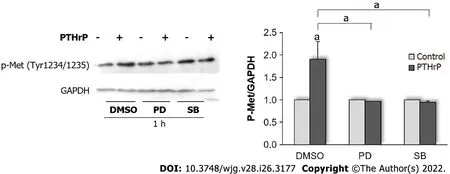
Figure 3 Parathyroid hormone-related peptide modulates Met activation through the mitogen-activated protein kinase signaling pathway in HCT116 cells. Cells were treated with PD-98059 or SB-203580, a selective mitogen-activated protein kinase (MAPK) kinase (MEK) or P38 MAPK inhibitor,respectively, for 30 min and then exposed to 10-8 mol/L PTHrP for 1 h to evaluate whether ERK1/2 MAPK and/or p38 MAPK mediate the effect of PTHrP on Met activation in HCT116 cells. Controls were run by adding an equivalent volume of dimethylsulfoxide, the vehicle of the inhibitors. The protein levels of Met phosphorylated in the residues Tyr1234 and Tyr1235 were assessed by Western blot. These phosphorylation sites constitute activating domains of the receptor.GAPDH protein levels were determined as a control of the amount of proteins present in the membrane, since this protein is not substantially modified with the treatment by the cytokine. Graph bars represent the average of the results obtained from three independent experiments. DMSO: Dimethylsulfoxide; GAPDH:Glyceraldehyde 3-phosphate dehydrogenase; Met: Receptor tyrosine kinase Met; p-Met: Phospho-Met (Tyr1234/1235); PTHrP: Parathyroid hormone-related peptide.aP < 0.05.
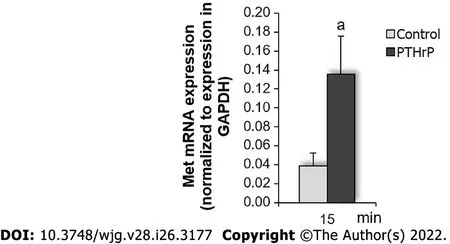
Figure 4 Parathyroid hormone-related peptide increases mRNA levels of Met in HCT116 cells. Colon cancer cells were exposed to parathyroid hormone-related peptide (PTHrP) for 15 min, followed by real-time polymerase chain reaction analysis to detect Met mRNA levels as described in Materials and Methods to evaluate whether PTHrP modulates Met mRNA levels in HCT116 cells. Graph bars represent the average of the results obtained from three independent experiments. GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; Met: Receptor tyrosine kinase Met; PTHrP: Parathyroid hormone-related peptide. aP < 0.05.
It has been shown that the loss of the expression of the epithelial marker E-cadherin is associated with EMT progress in CRC[76,77]. In our recent work[24], we showed that in HCT116 cells, PTHrP at 5 h modulated the expression of E-cadherin and other epithelial markers as well as certain mesenchymal markers. Based on these morphological changes related to the EMT and mediated through Met, we then explored by Western blot analysis whether Met is involved in the modulation of E-cadherin protein expression by the cytokine. As seen in Figure 5C, the effects of the cytokine on E-cadherin protein levels were reverted when the cells were pre-incubated with SU11274. Taken together, these findings suggest that PTHrP induces the EMT program in HCT116 cells through the Met signaling pathway.
In agreement with other works published[78], our findings suggest that the Met signaling pathway induced by PTHrP is involved in cell events related to tumor aggressive behavior in the HCT116 cells.
PTHrP attenuates the cytotoxic effect of CPT-11, OXA, and DOXO in human HCT116 cells through the Met signaling pathway
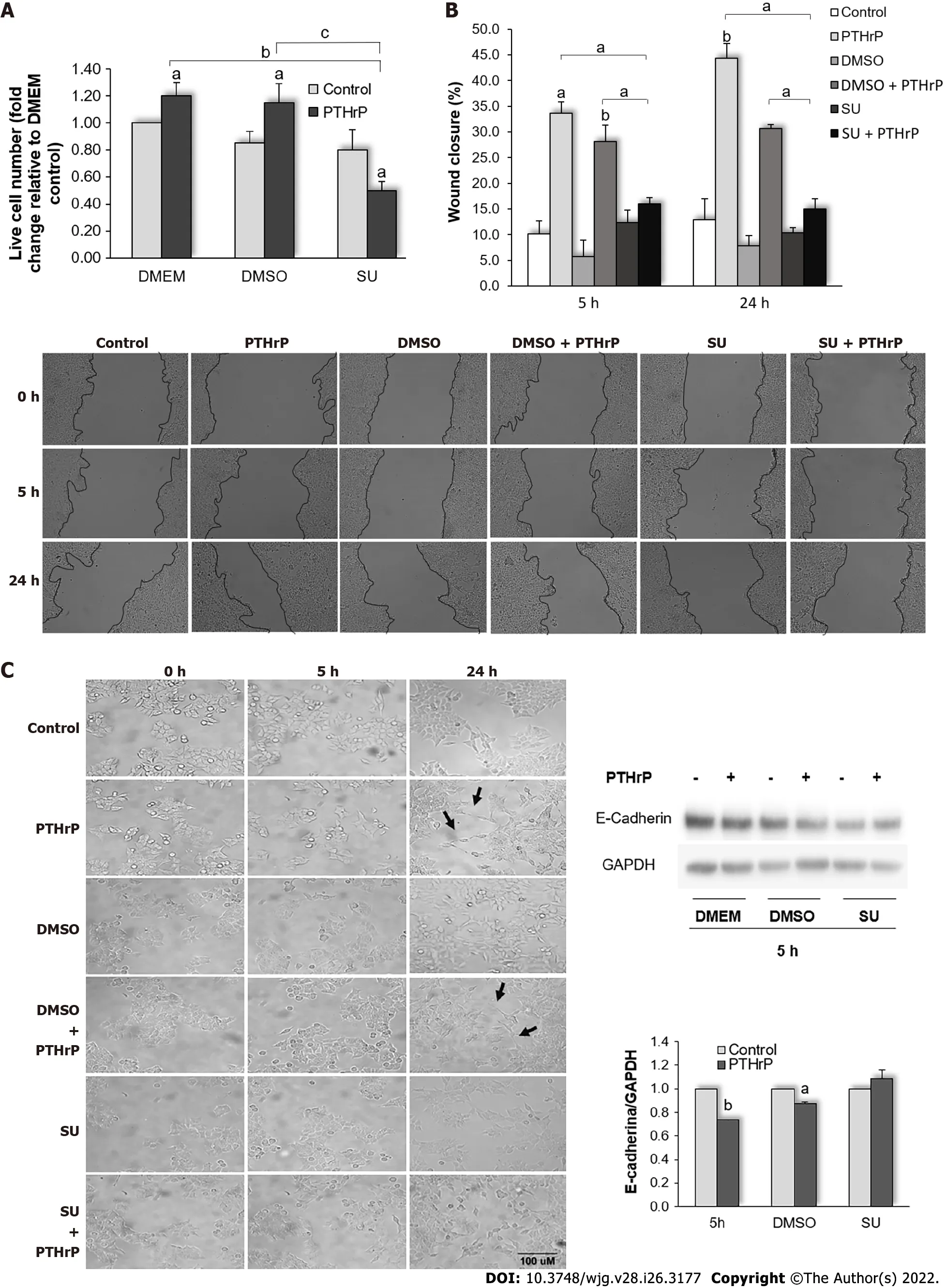
Figure 5 Parathyroid hormone-related peptide promotes events related to the aggressive behavior of HCT116 cells through the Met signaling pathway. HCT116 cells were pre-incubated with SU11274, a specific Met inhibitor, for 30 min and then treated with or without parathyroid hormonerelated peptide (PTHrP; 10-8M). A: Trypan blue technique showed that Met inhibition decreased the cell proliferation induced by PTHrP at 24 h; B: Images from wound healing assay show that Met inhibition reverted the wound closure promoted by PTHrP at 5 h and 24 h; C: E-cadherin protein levels analyzed by Western blot to investigate whether Met is involved in the decrease of this epithelial-mesenchymal transition (EMT) program marker induced by PTHrP in HCT116 cells. Using the Image J-NIH program, we performed the analysis of the parameters related to cell morphology. The arrows indicate the morphological changes corresponding to the transition from a polygonal structure to spindle-like structure related to EMT program progress observed when the cells were treated for 24 h with PTHrP or PTHrP plus dimethylsulfoxide (DMSO). In all experiments, a control with DMSO, the vehicle of the inhibitor, was performed. Graph bars represent the average of the results obtained from two independent experiments. DMEM: Dulbecco’s Modified Eagle Culture Medium; DMSO: Dimethylsulfoxide; EMT: Epithelial to mesenchymal transition; PTHrP: Parathyroid hormone-related peptide. aP < 0.05; bP < 0.01; cP < 0.001.

Figure 6 Analysis of the aspect ratio (major/minor axis) of HCT116 cells, obtained using the lmage J-NlH program. The table shows values of parameters associated to cell morphology: Area; perimeter; round; major axis; minor axis; and aspect radio, relation between major axis and minor axis (AR). Graph bars show the AR for each condition. The increase of the degree of cell elongation at 24 h of exposure to parathyroid hormone-related peptide was significantly reversed in the presence of the Met inhibitor. DMSO: Dimethylsulfoxide; PTHrP: Parathyroid hormone-related peptide; SU: SU11274. bP < 0.01.
In CRC-derived cells, our group has previously described a role of PTHrP in the resistance to CPT-11,the most widely used chemotherapeutic drug today[26]. In the treatment of CRC, many pharmacological strategies have been implemented. OXA is another drug commonly employed for this purpose and it exerts its cytotoxic action through different mechanisms with respect to CPT-11. Thereby, the combination of CPT-11 and OXA is generally used to improve the effectiveness of the adjuvant therapy[79,80]. DOXO is another chemotherapeutic agent that has been effective in the treatment of advanced CRC; however, the side effects associated with its use at high doses and the development of chemoresistance constitute a great challenge to the effective treatment[43,81,82]. Since the greatest problem in CRC treatment is the development of chemoresistance, we proceeded to evaluate if PTHrP affects the cytotoxicity promoted by these other drugs. Trypan blue dye exclusion test revealed that PTHrP not only decreased the sensitivity of intestinal tumor cells to CPT-11 but also to OXA and DOXO (Figure 7).
Studies carried out by colleagues demonstrated the correlation between overexpression and/or hyper-activation of Met in tumor tissues with a poor prognosis of cancer patients and with a chemoresistant phenotype[32,83,84]. These findings support the interest in Met inhibitors as new therapeutic strategies. Since herein we found that Met inhibition substantially reverts the effects of PTHrP on events associated with the malignant behavior such as cell proliferation, migration, and the EMT program, we set out to assess if the signaling pathway trigged by this RTK also promotes chemoresistance in our experimental system. Counting live cells by means of trypan blue dye exclusion test revealed that Met inhibition restored the sensitivity of HCT116 cells to CPT-11 (Figure 7A), OXA (Figure 7B), and DOXO(Figure 7C) even in the presence of PTHrP, suggesting that this cytokine attenuates the cytotoxic effect of these drugs in CRC cells through the Met signaling pathway.
PTHrP enhances the protein expression of Met and its receptor in HCT116 cells tumor xenografts
In a murine model, we have previously observed that the administration of PTHrP in HCT116 cell xenografts changed the protein expression of markers linked to CRC progression, such as Ki67, cyclin D1, ERK1/2 MAPK, CREB/ATF-1[26], RSK[27], VEGF[25], SPARC, and E-cadherin[24]. As Met overexpression promotes events related to tumor progression like proliferation, invasion, and migration[63,70]and taking into account our previous finding in the animal model, in this work we evaluated whether the administration of PTHrP in the same murine model (HCT116 cell xenografts in nude mice N:NIH(S)_nu) also changes the protein levels of Met in tumor tissues. To that end, 12 male mice, between 4 and 6 wk of age and with a body weight between 20 g and 25 g, were randomly divided into two groups (n= 6 each), after an acclimatization period of 4 d. Group 1 received the cytokine vehicle (100 μL PBS), while group 2 received PTHrP at a concentration of 40 μg/kg body weight in 100 μL PBS[49]. The treatment was intra-tumoral daily and until the end of the experiment to maintain the level of the compound constant in the tumor area. The mice were kept throughout the protocol in sterile conditions(see Materials and Methods). The immunohistochemical analysis with the quantification of Met immunoreactivity revealed that Met levels were elevated in tumors from mice treated with PTHrP(0.190 ± 0.014) compared to tumors from control mice (0.110 ± 0.012;P< 0.05) (Figure 8, top images). As the present work was aimed to elucidate the relationship between PTHR1, PTHrP, and Met, we also analyzed the immunoreactivity of PTHR1 using an anti-PTHR1 antibody in HCT116 cell xenografts from untreated nude mice (control) and mice treated with PTHrP. Interestingly, as seen in Figure 8(lower images), PTHrP increased the protein expression of its receptor in the tumor of these animals(2.27 ± 0.20) compared to tumors from control mice (1.98 ± 0.14;P< 0.01). The data shown in this section together with those in the first and fourth sections of results indicate that PTHrP modulates the expression of Met bothin vitroandin vivo. Perhaps the cytokine modulates the expression of its own receptor in this animal model in order to amplify its signaling within the cell and thus makes its effects more effective in CRC. More studies are necessary to confirm this hypothesis.
Evaluation of Met and PTHR1 expressions in CRC human samples
As we mentioned before, the Met receptor has the HGF as its only known ligand. However, it could be aberrantly activated by G-protein-coupled receptors[29,31]. The results reported herein in the previous sections suggest that PTHR1 activation (after its binding with PTHrP) is able to trigger the activation of the Met signaling pathway through a cross-talk between both receptors that is mediated by cytosolic kinases like Src. In this context, and taking into account the findings obtained byin vitroandin vivoassays, we decided to validate our observations and therefore our ultimate objective was to assess the expression of both receptors in CRC human tumor samples with the aim to elucidate if there is a relation between them and with the tumor characteristics. So, we analyzed 23 specimens obtained from patients with colorectal adenocarcinoma. Seven cases with normal colorectal tissues were assigned to the control group. Table 1 shows the patients’ characteristics. The average age was 62 years; 48% (n= 11) were male, and 52% (n= 12) were female. Regarding the primary tumor location, 39% (n= 9) presented rightcolon cancer, 30.5% (n= 7) presented left-colon cancer, while in 30.5% (n= 7) of patients the location was in the rectum; 61% (n= 14) of the tumors presented a high grade of histological differentiation, while 39% (n= 9) were moderately or poorly differentiated; 48% (n= 11) of the patients with CRC presented stage II, 39% (n= 9) stage III, and 13% (n= 3) stage IV.
The expression of Met and PTHR1 was evaluated in biopsies of patients with well (G1), moderately(G2), and poorly differentiated (G3) colon adenocarcinomas, as well as in normal colon tissue using the immunohistochemical technique. Figure 9A shows the comparison between both quantifications, Met and PTHR1, for each histological grade of differentiation. As shown in Figure 9B, in normal tissue Met receptor labeling is homogeneous throughout the cytoplasm of the epithelial cell. However, in tumor cells, we observed that the intensity of the immunostaining in the membrane increased as tumor differentiation declined. Concerning PTHR1, the intensity of the staining gradually decreased in histologically less differentiated tumors and the receptor seemed to relocate from the plasma membrane of the epithelial cell in G1 to the cytoplasm and the perinuclear zone in G3. Based on these observations, wedecided to investigate whether there is a correlation between the expression of Met and PTHR1. No statistical significance was found between the expression of both receptors in the tumor samples.Additionally, we studied if there is a statistically significant association between the clinicopathological characteristics and the immunohistochemical staining for Met or PTHR1. For this purpose, we dichotomized the specimens into low, medium, and high expression levels of Met or PTHR1. We found that in less differentiated tumors, Met expression increased (P= 0.035), while PTHR1 expression was statistically lower (P= 0.0496). However, we did not find a statistically significant association of Met or PTHR1 with age, sex, primary tumor location, and TNM stage. These results indicate that the expression of PTHR1 is more marked in well-differentiated tumors, while Met showed a higher expression level in poorly differentiated tumors.

Table 1 Clinicopathological characteristics of the patients
DlSCUSSlON
Resistance to chemotherapeutic drugs constitutes an obstacle in the therapy of CRC[2]. In this context, it is crucial to identify the molecular mechanisms related to the aggressive phenotype of CRC to develop more effective therapeutic strategies and find new prognostic and predictive markers.
PTHrP is expressed in various types of cancer, including CRC[16]. In fact, 95% of colorectal tumors present elevated levels of this protein[18-20,85]. Our research group found that this cytokine modulates different signaling pathways and events associated with the aggressive phenotype in CRC-derived cells[13,24-26].
Several studies have demonstrated the participation of the Met signaling pathway in the evolution of different types of cancer[28,33-35,83]. Moreover, this receptor is overexpressed and/or can be aberrantly activated by several mechanisms in CRC cells, triggering tumor development and progression[28,36,38,59]. A recent study addresses the relationship between the HGF/Met axis and cytokines such as PTHrP in bone metastasis and renal cell cancer progression[86]. In agreement with these observations, the results from this work suggest that the expression and activity of Met are regulated by signaling pathways trigged by the binding of PTHrP to PTHR1. We observed that exogenous PTHrP treatment modulates Met protein and gene expression in HCT116 cells. Despite that the cytokine regulates Met expression in HCT116 cells, our initial observations in Caco-2 cells reveal that PTHrP is not able to modulate its protein levels. In line with these findings, we previously reported that PTHrP modulates the protein levels of SPARC (an invasion marker) and E-cadherin (a marker related with EMT program)in HCT116 cells but not in Caco-2 cells[24]. The fact that the cytokine can modulate the expression of markers associated with the malignant behavior in a more aggressive cell line (HCT116) but not in a less aggressive cell line (Caco-2) may be because these CRC cells have different mutations[87,88] and perhaps several events triggered by PTHrP in these two cell lines may be dependent on these mutations explaining the different response between Caco-2 and HCT116 cells.

Figure 7 Parathyroid hormone-related peptide attenuates the cytotoxicity of CPT11, oxaliplatin, and doxorubicin through the Met signaling pathway. A-C: HCT116 cells were pre-incubated with SU11274, a specific Met inhibitor, and then treated with PTHrP (10-8 M) and/or irinotecan (CPT-11,10 μM) (A), oxaliplatin (OXA, 10 μM) (B), or doxorubicin (DOXO, 5 μM) (C) for 24 h. The number of viable cells was quantified by Trypan blue technique. In each experiment, a control with DOXO, the vehicle of the inhibitor, was performed. Graph bars represent the average of the results obtained from two independent experiments. CPT-11: Irinotecan; DMSO: Dimethylsulfoxide; DOXO: Doxorubicin; OXA: Oxaliplatin; PTHrP: Parathyroid hormone-related peptide; SU: SU11274. aP <0.05; bP < 0.01; cP < 0.001.
We found in HCT116 cells that the peptide promotes the activation of Met by phosphorylation on Tyr residues. Bradley and colleagues and later Critchley and his research group, demonstrated that, like other RTKs, Met after its activation is degraded by the proteasomal pathway[62,63]. Consistent with these data, our results suggest that Met protein levels decline after PTHrP-induced phosphorylation.
Previous studies by our research group established that PTHrP in the HCT116 cell line promotes phosphorylation and activation of Src; then this kinase acts upstream of ERK 1/2 MAPK to induce their phosphorylation/activation, though the mechanisms that lead to the phosphorylation/activation of p38 MAPK by the cytokine is still unknown[13,26]. Herein, we demonstrated that PTHrP induces the phosphorylation and activation of Met through Src. Furthermore, PTHrP modulates the phosphorylation/activation of Met through ERK 1/2 and p38 MAPK. The fact that the cytokine diminishes Met protein levels in a range of time between 1 h to 5 h suggests that Met is activated by PTHrP (with its subsequent degradation) not only by the MAPKs but also by some pathway/s that is/are effector/s of the action of these MAPKs. This idea is supported by our finding that the effects of PTHrP is mediated by these MAPKs in HCT116 cells at longer exposure times[26]. More studies are needed to confirm this hypothesis.
Given these observations, the results obtained in this work suggest that at least the Src-ERK 1/2 MAPK and p38 MAPK axis is required for the phosphorylation/activation of Met induced by PTHrP in our model. Since MAPKs are serine-threonine kinases and Met requires phosphorylation on tyrosine residues for its activation, further studies are necessary to determine the link between these molecules.
As we previously mentioned, in CRC, the Met signaling pathway is associated with tumor evolution and also with resistance to chemotherapeutic drugs[32]. Currently, the inhibition of this RTK is being widely studied as a complementary therapy to conventional CRC treatments[37,38]. SU11274 prevents Met activation because it is an ATP competitive inhibitor of Met catalytic activity[70]. We previously showed that PTHrP has several effects on CRC derived cells such as proliferation, survival, migration,and induction of EMT program, among others[13,26,39]; the fact that we found a significant reduction in the viability and migration of HCT116 cells in the presence of an Met inhibitor as well as the reversal of the mesenchymal phenotype induction, even in the presence of PTHrP, indicates that Met mainly participates in the molecular mechanisms that are involved in these cell responses to PTHrP action.

Figure 8 Parathyroid hormone-related peptide increases the protein expression of Met and parathyroid hormone receptor type 1 in tumor xenografts. Met and parathyroid hormone-related peptide receptor type 1 (PTHR1) protein levels were evaluated by the immunohistochemistry technique in the HCT116 cell tumor-bearing nude mouse model. Tumor sections were stained with an anti-Met antibody or anti-PTHR1 antibody. Images (400 ×) are from the tumor treated with saline solution (left) or with PTHrP (right). The immunostaining was quantified with the Fiji image processing package of the Image J-NIH program. Scale bar: 50 μM. Met: Receptor tyrosine kinase Met; PTHR1: Parathyroid hormone receptor type 1; PTHrP: Parathyroid hormone-related peptide. aP < 0.05; bP < 0.01.
PTHrP favors the aggressive behavior of CRC cells, inducing other events related to the malignant phenotype such as chemoresistance to CPT-11. The tumor cell response to this drug under PTHrP action involves the ERK signaling pathway[26]. Other investigations carried out by Paillas and colleagues demonstrated that the p38 MAPK pathway also modulates the sensitivity of CRC cells to CPT-11[89].Given the data obtained previously by us and by other researchers and taking into account the link in CRC between Met signaling and the MAPKs pathway showed in this work and by other authors[41],our next motivation was to further investigate the chemoresistance of CRC-derived cells induced by PTHrP by analyzing two key aspects: Whether the cytokine affects the sensitivity of CRC cells to other chemotherapeutic drugs and whether the Met signaling pathway participates in PTHrP-trigged chemoresistance.
Our work shows that the treatment with this cytokine also attenuates the cytotoxicity induced by OXA and DOXO. This is an interesting result as it indicates that PTHrP promotes resistance to different types of cytotoxic agents. Perhaps the mechanisms trigged by this cytokine alter specific targets or the signaling of these drugs. Tumor cells develop different strategies to avoid chemotherapeutic drug effects. To improve the efficacy of the treatment, combinations of multiple agents are usually used[90].Given that these drugs present different modes of action, pharmacological doses, and side effects[91-93], in the future it will be necessary to continue this research by testing new drugs and combinations thereof as well as elucidating the specific targets of PTHrP for each one.
Besides, the use of SU11274 together with CPT-11, OXA, or DOXO increases the sensitivity of CRC cells to these drugs, suggesting that Met participates in the chemoresistance induced by PTHrP. The inhibitors of markers/pathways related to cancer progression are still under investigation and like all anti-cancer approaches, their applications have challenges in the therapy of the disease[78,94]. With respect to the inhibition of the Met signaling pathway, a recent publication by Du and colleagues proposes to target the HGF/Met axis as a possible strategy for patients with metastatic CRC[94].
So far, ourin vitroobservations have allowed us to suppose the existence of a mechanism based on the action of PTHrP on the regulation ofMetgene expression and also on its activation through Src kinase and the MAPKs pathway. Once activated, Met signaling leads to molecular changes in the tumor cell to promote chemoresistance to CPT-11, OXA, and DOXO. Probably, the up-regulation of Met expression also collaborates in the induction of events associated with the aggressive behavior of CRC cells (Figure 10). Besides, our results support those studies that suggest the incorporation of Met inhibitors combined with adjuvant drugs as new therapeutic strategies in CRC treatment and establish the need to further investigate whether PTHrP is a predictive and/or prognostic marker of CRC.

Figure 9 Relation of Met and parathyroid hormone receptor type 1 staining intensity with tumor histological differentiation. A: Quantification of the immunostaining by optical density analysis using the FIJI plug-in from the Image J program; B: Representative images (200 ×) obtained by hematoxylin-eosin staining and by the immunohistochemistry technique performed for Met and PTHR1 for normal colorectal tissue and colorectal cancer tissue, with histological grades G1, G2, and G3. Scale bar: 10 μM. H-E: Hematoxylin-eosin staining; Met: Receptor tyrosine kinase Met; NT: Normal tissue; PTHR1: Parathyroid hormone receptor type 1.
In this work, we also performedin vivoassays with the aim to evaluate the effects of PTHrP in a different context for tumor cells. As we expected, in agreement with ourin vitroobservations, HCT116 cell tumor xenografts treated with the peptide exhibited an increase in Met protein expression concerning the control. PTHrP increases the levels of its own receptor, perhaps to amplify its signaling within the cell, and thus makes its effects more effective in CRC. Despite the limitations of the N:NIH(S)_nu model regarding the interaction of tumor cells with the stroma[13,24], these results allow us to presume a relationship between the aberrant expression of Met already observed in the tumor tissue of CRC patients[38] and PTHrP/PTHR1.
As we previously stated, although HGF is the only known ligand capable of inducing Met phosphorylation and activation, studies have shown that the cross-communication with G-coupled protein receptors transactivates this RTK. Concerning this, it was demonstrated that the prostaglandin E2 receptor EP1 can induce an aberrant activation of RTKs like Met in hepatocellular carcinoma cells[30]. Similar results had previously been obtained by Fischer and colleagues in pancreatic and hepatocellular carcinoma cells[29]. As we previously mentioned, PTHrP has intracrine, autocrine, endocrine,and mainly paracrine actions through the binding to a G-coupled protein receptor, PTHR1[9,10]. For this reason, the peptide and its receptor are expressed in the same cells or adjacent cells and changes in their expression are directly correlated[14,15]. Considering the previously described background, we evaluated Met and PTHR1 expression in normal colorectal tissues and in biopsies from patients with colon adenocarcinoma. Although Lee and colleagues did not find a statistical relevance between Met overexpression and the clinicopathological characteristics of patients with CRC, they demonstrated that it is associated with a lower overall survival and progression-free survival[38]. In contrast, Kim and his research group concluded that Met expression in patients with CRC is not a prognostic indicator for overall survival[95]. Despite these data, there was no information about the association between Met and PTHR1 expression and its impact on the evolution of CRC. As we expected, given that both receptors have a physiological role, Met and the PTHrP receptor are expressed in normal colon tissue.We found a statistically significant result when we compared the staining of tumors with high histological differentiation compared to those more undifferentiated with regard to the immunoreactivity for Met and PTHR1 that gradually increase and diminish, respectively.
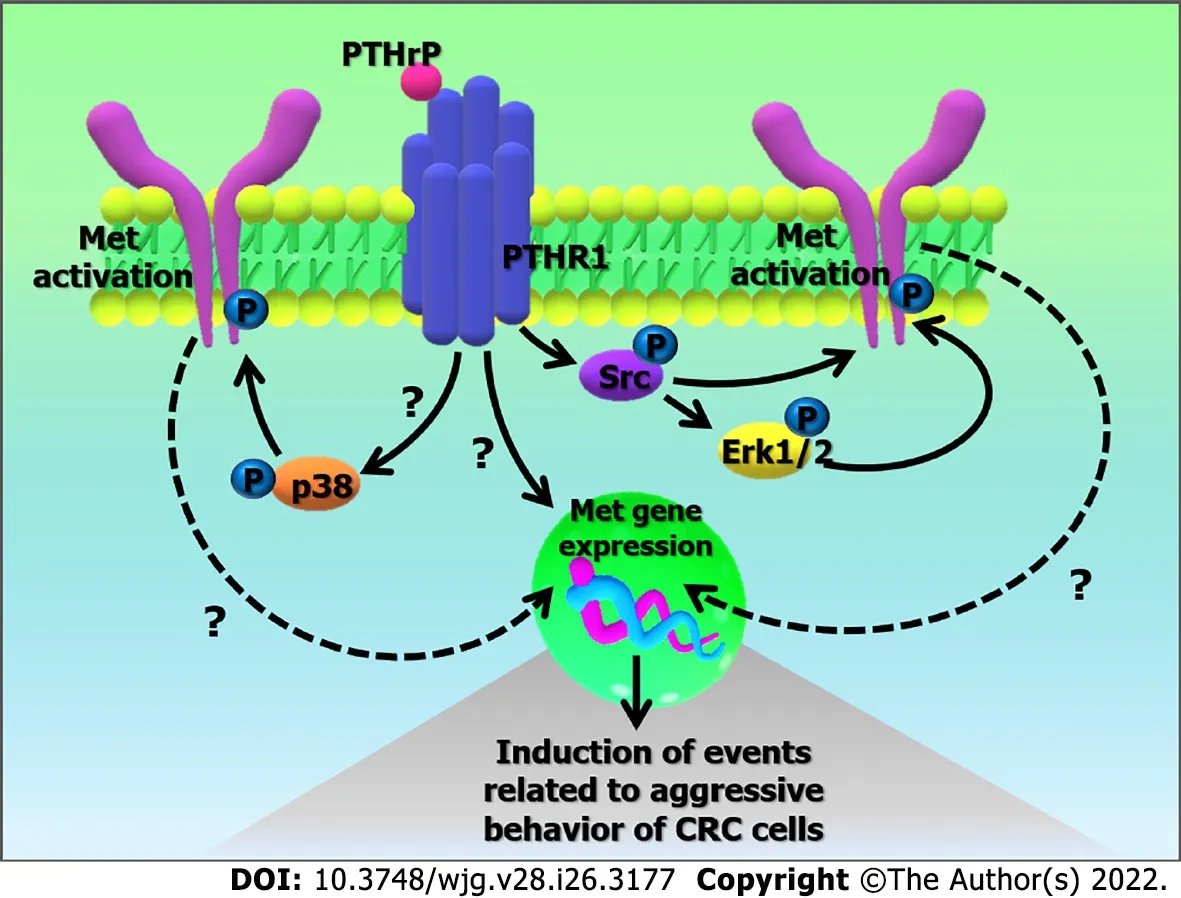
Figure 10 Possible mechanism based on the action of parathyroid hormone-related peptide in the regulation of Met gene expression and its activation. The binding of parathyroid hormone-related peptide (PTHrP) to PTHR type 1 (PTHR1) in HCT116 cells promotes the up-regulation of Met gene expression. PTHrP also induces the activation of Met through Src kinase and the mitogen-activated protein kinases (MAPKs) pathway (Erk 1/2 MAPK and p38 MAPK). Once activated, Met signaling leads to molecular changes in the tumor cell that promote events related to the aggressive behavior of colorectal cancer cells.CRC: Colorectal cancer; Erk 1/2: Extracellular signal-regulated kinases 1/2; Met: Receptor tyrosine kinase Met; P: Activators domains phosphorylated; PTHR1:Parathyroid hormone receptor type 1; PTHrP: Parathyroid hormone-related peptide.
Interestingly, García and colleagues showed that in neuroblastoma, PTHrP acts as a growth factor promoting tumor progression and high levels of its receptor are associated with less aggressive tumor characteristics; also, the intracrine and paracrine actions of the cytokine promote phenotypes with different levels of aggressiveness in their experimental models. These researchers found that PTHR1 is poorly expressed in neuroblastoma cell lines that are not well differentiated; however, in those cells with a high degree of differentiation, PTHR1 has greater expression. Furthermore, they observed that the receptor knockdown promotes a much more aggressive phenotype in more differentiated cells[96].These data support our observations and could be related with the role of PTHrP and PTHR1 in the promotion of an aggressive phenotype of CRC.
We observed that the intensity of Met immunostaining in tumor cell plasma membrane increases as tumor differentiation declines. Concerning the location of PTHR1 in the plasma membrane, the intensity of the staining gradually decreases in histologically less differentiated tumors; also, it is located in the cytoplasm and the perinuclear region of the tumor cell, with immunoreactivity more predominant in samples with histological degree G3. This could be related to an intracrine role of the cytokine most preponderant in less differentiated CRC cells. More studies are necessary to confirm this.
Based on the above, it is presumed that in the early stages of tumor development, PTHrP would act by binding to its receptor in the tumor cell, possibly inducing Met receptor expression. The secretion of this cytokine from tumor stromal cells has also been reported[97] and we have previously demonstrated its influence on the synthesis and release of tumor microenvironment factors that favor an aggressive CRC phenotype[24,25]. Considering that in less differentiated tumors, we observed lower expression of PTHR1, we hypothesize that in this instance the tumor stromal cells adopt a leading role by secreting(perhaps in response to PTHrP) other cytokines that promote the expression and activation of molecular markers related to a worse prognosis of the disease. The high levels of Met in more advanced stages could be mediated not only by the action of PTHrP but also by other cytokines as other researchers have previously demonstrated[63,98]. It is known that the loss of cellular identity, characteristic of an advanced undifferentiated state, is evidenced by changes in the expression of surface molecules including receptors on tumor cells[99]. The high levels of PTHR1 observed in normal samples and primary neoplasms, and its decrease in undifferentiated histological grades could respond to this effect.On the other hand, high expression of Met in tumor samples is observed in association with a low degree of cellular differentiation. In this context, perhaps PTHR1 and Met could act as markers of cellular dedifferentiation. However, more studies are needed to verify this hypothesis.
CONCLUSlON
Here we have demonstrated using anin vitromodel that the PTHrP pathway promotes events that are associated with a worse evolution of CRC through Met activation. We also provide evidence for the association between PTHrP action and Met expressionin vivo. Given all this information, it will be of great interest to ascertain the participation of Met in other molecular events related to CRC progression trigged by PTHrP and its translational relevance. More studies are needed in the areas of basic and clinical oncology to confirm this.
ARTlCLE HlGHLlGHTS


FOOTNOTES
Author contributions:Novoa Díaz MB and Carriere P contributed to conceptualization, methodology, investigation,formal analysis, visualization, and manuscript drafting, review, and editing; Gigola G and Zwenger AO contributed to conceptualization, methodology, and investigation; Calvo N contributed to conceptualization, methodology,investigation, formal analysis, visualization, supervision, and manuscript drafting, review, and editing; Gentili C contributed to conceptualization, methodology, resources, investigation, formal analysis, visualization, supervision,manuscript drafting, review, and editing, project administration, and funding acquisition.
Supported bythe Agencia Nacional de Promociόn Científica y Tecnolόgica, No. PICT-2013-1441; Consejo Nacional de Investigaciones Científicas y Técnicas, No. PIP11220150100350; Instituto Nacional del Cáncer Asistencia Financiera II,RESOL 493/14, No. 2002-4395-14-1; Instituto Nacional del Cáncer Asistencia Financiera III-2016-2017, RESOL-2016-1006-E-APN-MS, No. 2002-3862-16-1 CANCER; Universidad Nacional del Sur, No. PGI: 24/B230 and No. PGI:24/B303; and Fundaciόn Alberto J. Roemmers of Argentina.
lnstitutional review board statement:The study was reviewed and approved by the committee of experts on the subject conformed by members of the Department of Biology, Biochemistry and Pharmacy of the National University of the South.
lnstitutional animal care and use committee statement:All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All experiments with animals were approved by a local animal committee for ethics (CICUAE-UNS, institutional endorsement updated to 2021), and were carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH 1996).
Conflict-of-interest statement:There are no conflicts of interest to report.
Data sharing statement:No additional data are available.
ARRlVE guidelines statement:The authors have read the ARRIVE Guidelines, and the manuscript was prepared and revised according to the ARRIVE Guidelines.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Argentina
ORClD number:María Belén Novoa Díaz 0000-0002-6684-4076; Pedro Carriere 0000-0002-5751-8850; Graciela Gigola 0000-0001-7362-3178; Ariel Osvaldo Zwenger 0000-0002-4407-5413; Natalia Calvo 0000-0003-3627-6749; Claudia Gentili 0000-
0002-9957-7824.
S-Editor:Chen YL
L-Editor:Wang TQ
P-Editor:Yu HG
 World Journal of Gastroenterology2022年26期
World Journal of Gastroenterology2022年26期
- World Journal of Gastroenterology的其它文章
- Role of gadoxetic acid-enhanced liver magnetic resonance imaging in the evaluation of hepatocellular carcinoma after locoregional treatment
- Clinical implications and mechanism of histopathological growth pattern in colorectal cancer liver metastases
- Bifidobacterium infantis regulates the programmed cell death 1 pathway and immune response in mice with inflammatory bowel disease
- Tumor-feeding artery diameter reduction is associated with improved short-term effect of hepatic arterial infusion chemotherapy plus lenvatinib treatment
- Impact of sodium glucose cotransporter-2 inhibitors on liver steatosis/fibrosis/inflammation and redox balance in non-alcoholic fatty liver disease
- Endoscopic techniques for diagnosis and treatment of gastro-entero-pancreatic neuroendocrine neoplasms:Where we are