
基于重测序的短节间番茄品系变异检测分析
2022-07-29 09:20李毅丰姜悦畅孙中锋唐贝贝师海林王玉斌毛秀杰
江苏农业科学 2022年13期
李毅丰, 姜悦畅, 孙中锋, 王 帅,2, 唐贝贝, 师海林, 张 宁,2, 王玉斌, 毛秀杰,2
(1.河北科技师范学院园艺科技学院,河北秦皇岛 066004;2.河北省特色园艺种质挖掘与创新利用重点实验室,河北秦皇岛 066004; 3.河北省承德市农业农村局,河北承德 067000)
番茄(L.)是重要的蔬菜作物和模式植物。实际生产应用中,集约化的育苗方式增加了番茄育苗密度,植株间相互遮阴,促使幼苗发生徒长,因此番茄节间长度的研究受到广泛关注。番茄节间长度是影响番茄产量的重要农艺性状之一,不仅会对番茄栽培密度产生影响,还对番茄的叶面积指数、光能利用率、干物质积累以及果实品质产生影响。番茄节间长度缩短不仅能够有效降低植株高度,抑制徒长,增加土地利用率,减少土壤中营养物质的消耗,还能降低苗期管理和果实采收的人工成本,因此,对短节间番茄进行研究具有重要意义。
随着测序技术的迅速发展,通过高通量测序技术能够对许多作物基因进行研究。周世奇等对选育的航天突变体烟草NC89-M与野生型NC89进行全基因组重测序,在NC89中检测获得了1 848 013个单核苷酸多态性(single nucleotide polymorphism,SNP)、398 922个小片段插入和缺失(insertion-deletion,InDel)、41 969个染色体结构变异(structural variation,SV),在NC89-M中检测到 1 876 219 个SNP,402 011个InDel,42 699个SV,采用生物信息学分析方法得出代谢通路和次生代谢产物合成相关基因突变数目最多,通过变异基因功能注释获得-基因和基因,分别调控开花时间和侧生器官发育与叶缘形状。杜海东等对3个自封顶番茄品系进行基因组重测序,通过变异检测分析3个样本检测到 5 968 501 个SNP和485 114个InDel,与参考基因组比对后共发生33 473个变异基因。对CDS区域的变异基因进行KEGG数据库比对,发现主要集中在基础代谢和玉米素的生物合成,通过多序列比对获得了16个调控封顶花序数的关键基因。
目前,关于番茄节间长度的研究,多集中于对长节间番茄的研究,而对短节间番茄突变体的研究鲜见报道。因此,本研究对2个节间长度不同的番茄品系进行全基因组重测序,并对其SNP、InDel、拷贝数变异(copy number variation,CNV)、SV等4种变异类型进行深度挖掘,以及基因变异对代谢通路的影响,从而为分子水平改良番茄节间长度的研究奠定基础。
1 材料与方法
1.1 试验材料
本试验以短节间番茄品系CH和普通节间番茄品系DH为试验材料,均为无限生长型。试验在河北科技师范学院园艺园林试验站7号温室中进行,于2021年1月16日进行播种育苗,待幼苗长至5~6张真叶时进行移栽。采用日光温室栽培,大行距为70 cm,小行距为40 cm,株距为30 cm,应用滴灌方式进行水肥管理。在开花结果期取幼嫩组织用于基因组重测序分析。
1.2 试验方法
1.2.1 全基因组DNA提取 通过十六烷基三甲基溴化铵(CTAB)法对样本茎段进行DNA的提取;用0.8%琼脂糖凝胶电泳对DNA质量进行检测;采用核酸蛋白分析仪对提取的DNA进行定量。
1.2.2 全基因组重测序 对质检合格的DNA采用超声进行随机打断。采用TruSeq DNA PCR-Free Prep kit的标准建库流程制备测序文库,对序列末端进行修复、3′端添加多聚腺苷酸(PloyA)、5′端添加含有文库特异性标签、纯化、测序文库模板富集等步骤完成测序文库的制备。对完成的测序文库在Agilent Bioanalyzer上进行质检;采用Quant-iT PicoGreen dsDNA Assay Kit对文库进行定量(文库浓度大于2 nmol质量合格);根据所需测序量对合格的各上机测序文库梯度稀释后按相应比例混合使用;应用NovaSeq测序仪进行双端测序。
1.2.3 数据统计 采用BWA(0.7.12-r1039)mem程序将经过过滤后得到的高质量数据比对到参考基因组上,比对的参数均按照bwamem的默认参数。采用ANNOVAR软件对SNP位点和InDel位点进行注释。采用GATK(https://www.broadinstitute.org/gatk/)(RealnTimes = 1)进行CNV检测,使用BreakDancer进行结构变异(SV)检测。
2 结果与分析
2.1 CH与DH农艺性状分析
在开花结果期对2个番茄品系的株高、总节间长度、平均节间长度进行分析(表1、图1)。结果表明,CH株高均极显著高于DH;CH与DH相比总节间长度不存在显著差异;CH与DH之间的平均节间长度均存在极显著差异。

表1 CH与DH开花结果期农艺性状分析

2.2 变异检测分析
2.2.1 CH与DH基因组重测序结果与分析 由表2可见,CH和DH这2个样本的Reads总数分别为 168 148 334、152 257 248个,过滤后Reads数量及过滤后Reads数量占原始Reads数量的百分比分别为160 322 604个(95.35%)、145 443 866个(95.53%);碱基总数分别为25 222 250 100、22 838 587 200 bp,过滤后Reads碱基数及高质量Reads碱基数占原始碱基总数的百分比分别为 23 522 020 839 bp(93.26%)、21 329 453 341 bp(93.39%)。GC含量在36.83~36.98之间;Q20≥96.82%、Q30≥91.85%。

表2 CH与DH的碱基数据统计
2.2.2 CH和DH与参考基因组对比情况 以Heinz 1706为参考基因组。2个样本的比对率在99.61%~99.89%之间(表3)。平均测序深度为23X,1X覆盖度在96.63%~99.74%之间,4X 覆盖度在95.62%~99.52%之间,10X 覆盖度在93.19%~96.97%之间,20X 覆盖度在46.47%~52.31%之间(表4)。综上,比对结果正常,可用于后续相关分析。
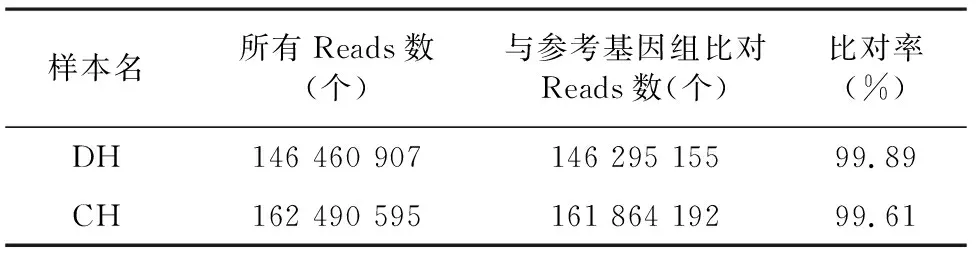
表3 CH与DH序列比对结果统计

表4 CH与DH比对测序深度和覆盖度结果统计
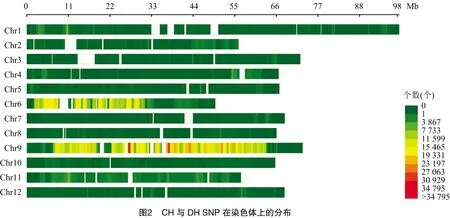
2.2.3 SNP检测与注释 依据与参考基因组的比对结果(表5)可知,CH和DH的总SNP变异个数相同,为3 893 609个,CH和DH的纯合基因型数分别为186 147、3 506 620个,CH中纯合基因型低于DH;杂合基因型数分别为1 106 264、251 093个;未知基因型数分别为36 007、2 994个;与参考基因组不一致的纯合基因型数分别为 2 565 191、132 902个。由SNP在染色体上的分布(图2)可知,SNP在9号染色体的24.89~27.79 Mb、35.32~38.21 Mb区域内富集,初步判断在此区域内发生基因突变,其余SNP均匀分布在6号染色体的1.74~8.11 Mb、12.16~31.26 Mb和9号染色体的7.53~18.53 Mb、21.42~62.53 Mb之间。
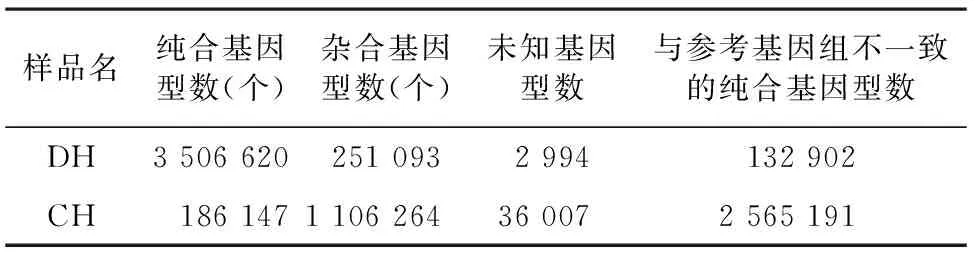
表5 CH与DH SNP检测结果统计
依据参考基因组比对结果,对CH和DH中的SNP进行比较(表6),结果表明,CH和DH中共检测到1 086 531个SNP。对2个样本之间的全基因组SNP变异进行注释,发现SNP变异主要集中在基因间区,其比例约占总数的78.03%;其次是发生于内含子区域内,其比例约占总数的8.48%;而发生于CDS区域的非同义突变比例约占CDS区域总数的60.85%。
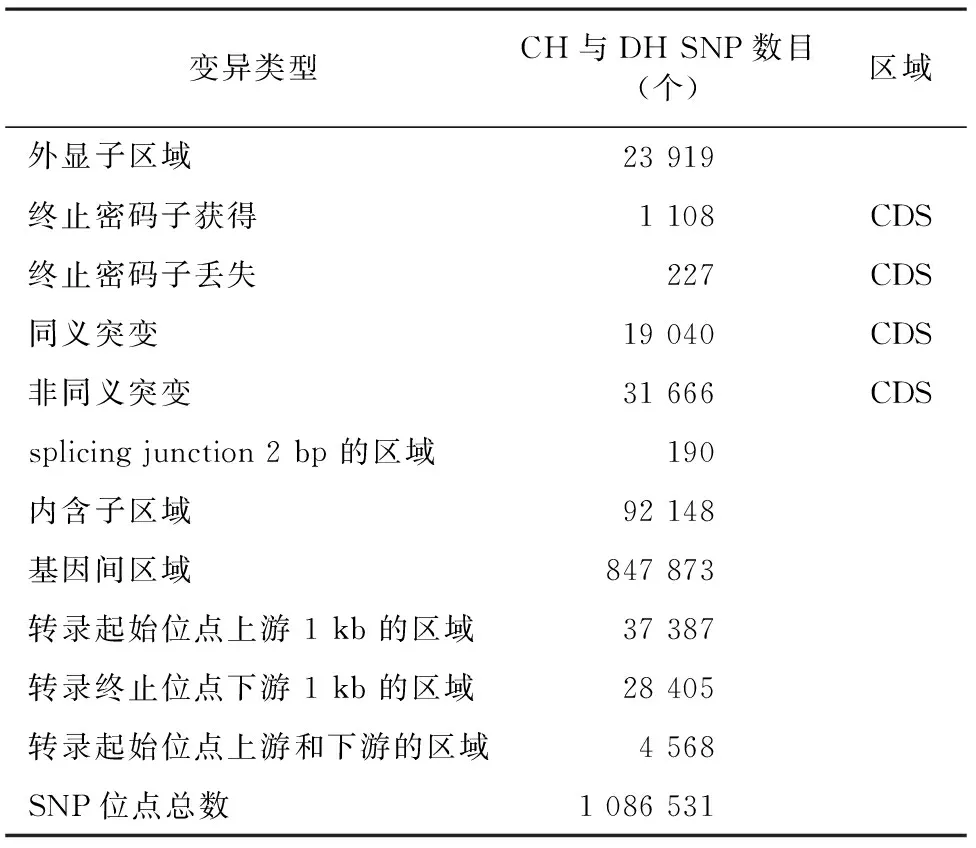
表6 CH与DH SNP注释结果
基因组SNP突变可以分成6类,分别为T:A>C:G、T:A
2.2.4 InDel检测及注释 为了定位目标性状,每组性状相关样本一起call群体InDel,经过滤对最终得到的群体SNP在每个样本中的数目做统计见表7。由表7可知,CH和DH的纯合基因型数分别为15 327、259 512个,CH中纯合基因型数小于DH;杂合基因型数分别为76 637、29 143个,CH中杂合基因型数多于DH;未知基因型数分别为3 180、733个;与参考基因组不一致的纯合基因型数分别为265 384、71 140个。
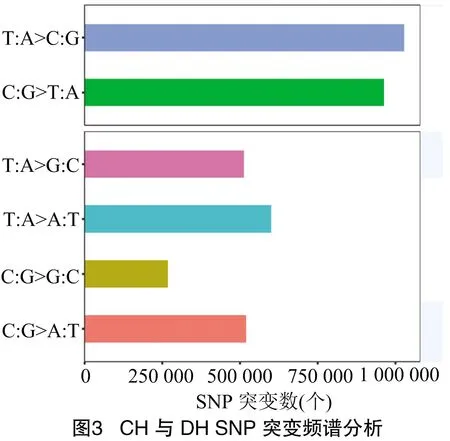

表7 CH与DH InDel检测结果统计
为鉴定CH与DH之间的InDel变异,分别进行比较(表8)。结果表明,CH和DH之间共检测到277 973个InDel。对2个样本之间的全基因组InDel变异进行注释,发现InDel变异主要集中在基因间区域,其比例约占总数的76.53%;其次是发生于内含子区域内,其比例约占总数的11.67%;而发生于CDS区域的移码突变比例约占总数的0.55%。
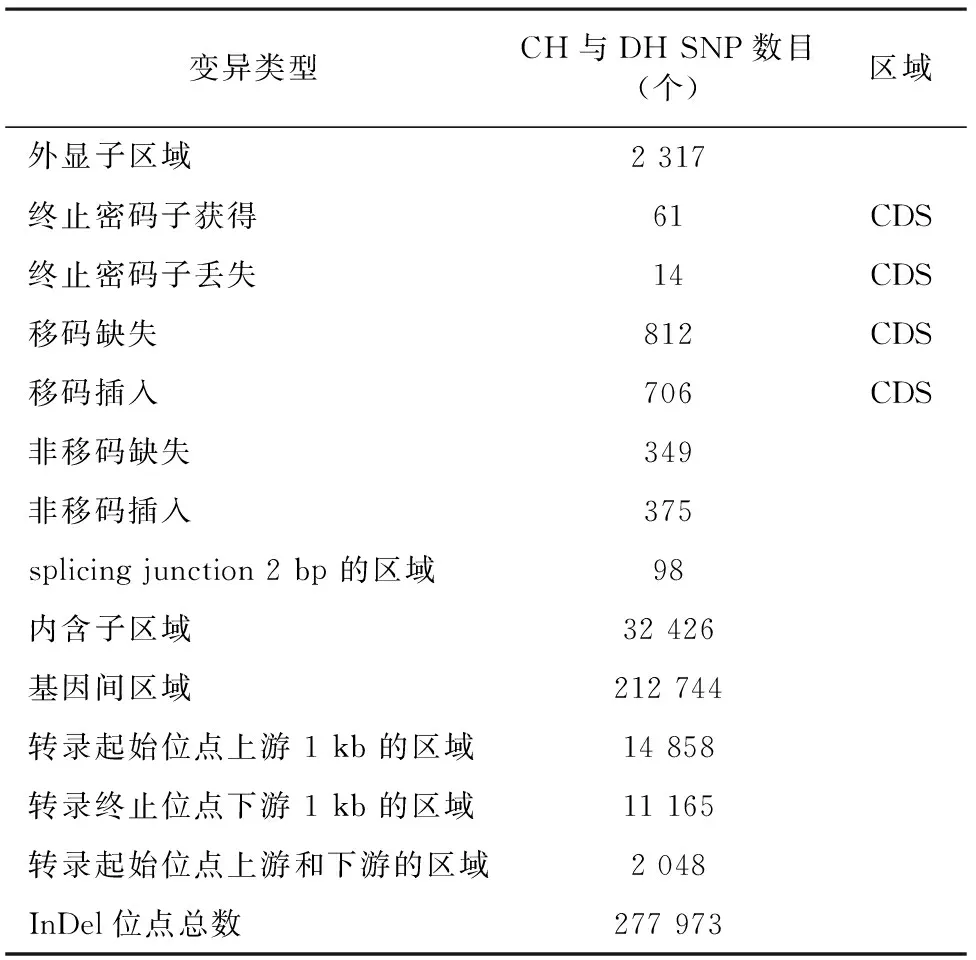
表8 CH与DH InDel注释结果统计
2.2.5 CH与DH CNV变异分析 采用CNVnator v0.2.7检测3个样本的全基因组中存在的CNV,通过CNV统计(表9)可知,CH中检测出的CNV为 12 756 个,拷贝数缺失10 970(85.9%),拷贝数增加1 786(14.1%);在DH中检测出的CNV为 13 041 个,拷贝数缺失11 576(88.8%),拷贝数增加1 465(11.2%)。因此得出,CH和DH的CNV变异主要是发生拷贝数缺失。
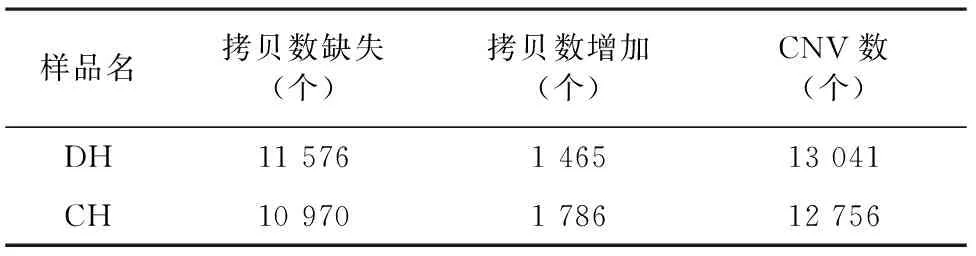
表9 CH与DH CNV变异分析
2.2.6 CH与DH SV变异分析 染色体的结构变异主要包括缺失(deletion,DEL)、插入(insertion,INS)、倒位(inversion,INV)、染色体内易位(intra-chromosomal translocation,ITX)、染色体间易位(inter-chromosomal translocation,CTX)。采用Breakdancer1.3.7版本检测染色体结构变异,5种结构变异的数量统计见表10。CH的SV数量最多为20 501个,其中染色体缺失的数量最多为8 176个,占总变异的39.9%,染色体间易位和染色体内易位分别占总变异的24.6%、20.0%,插入和倒位分别占总变异的5.5%、10.0%。DH的SV数量最少为13 015个,其中变异最多的2种类型为染色体间易位和缺失分别为4 668个、4 468个,占总变异的35.9%、34.3%,插入、倒位、染色体内易位分别占总变异的3.7%、6.5%、19.6%。CH和DH的SV变异主要发生的变异类型为缺失。

表10 CH与DH SV变异分析
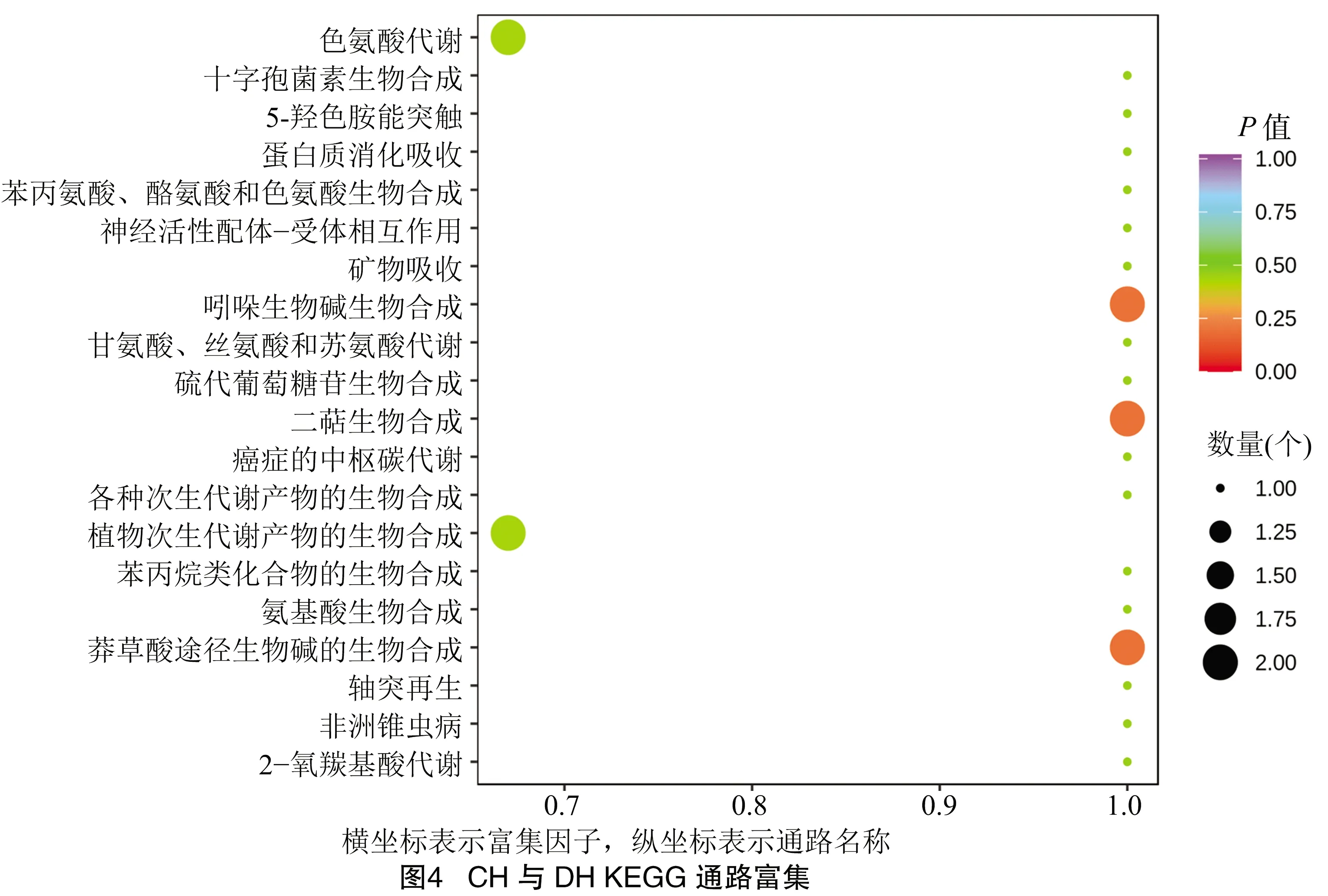
2.2.7 CH与DH代谢水平的差异分析 为了解基因变异引起代谢物质产生差异,因此对CH与DH节间中的代谢物质进行了分析。根据差异代谢物结果,对CH与DH的KEGG通路进行比较,获得KEGG通路富集图(图4)。由图4可知,在CH和DH中共注释到20个代谢通路,其中有3个通路被显著富集,包含吲哚生物碱生物合成、二萜生物合成、草莽酸途径生物碱的生物合成。
2.2.8 番茄短节间形成相关基因挖掘 为挖掘参与调控番茄短节间形成的相关基因,以SNP以及InDel中的基因作为切入点,进行变异基因挖掘。通过代谢通路富集情况,筛选出3个与节间长度相关联的变异基因,各基因功能注释、变异类型见表11,根据功能注释获得了控制赤霉素氧化酶、赤霉素受体、生长素响应因子的基因。

表11 变异基因功能注释
2.2.9 候选基因表达分析 基于qRT-PCR 技术检测开花结果期中与节间长度相关联的变异基因,并进行相对表达量分析。由图5可得,3个变异基因在2个试验材料中均表现出差异。基因在DH中的表达量是CH中的2.85倍;基因在DH中的表达量是CH中的5.87倍;基因在DH中的表达量是CH中的1.58倍。在DH中3个基因的表达量明显高于CH,以此推断、、在番茄节间长度的调控中发挥着至关重要的作用。

3 讨论与结论
番茄是重要的蔬菜作物,其节间长度受多种因素影响,包括环境因子、植物激素、遗传因子等。目前已有植物节间长度分子研究的报道,刘根忠等通过对226份番茄核心种质资源进行全基因组关联分析,鉴定出7个与节间长度相关的数量性状基因座(QTL),通过对显著的SNP位点进行候选基因分析,挖掘出控制番茄节间长度的主效基因和;刘忠祥等通过对玉米进行外源赤霉素(GA)的喷施,确定玉米对GA的响应正常,在利用分子标记技术将控制株高的主效QTL qPH3.2共定位在第3染色体上。研究发现,不同的土壤环境会对番茄内源激素含量产生影响,从而对植株的生长发育产生影响;控制节间发育相关的基因通过影响代谢通路来控制植物节间发育。前人研究发现,分子标记辅助育种能够提高植物优良性状的选择效率,对植物遗传育种具有重要意义。随着番茄全基因组测序的完成以及二代测序的迅速发展,使番茄基因组的变异检测分析成为可能。
本研究通过对短节间番茄品系CH和普通节间番茄品系DH进行全基因组重测序,与参考基因组Heinz 1706番茄相比,在DH中检测到 3 893 609 个SNP、360 528个InDel、13 041个CNV、13 015个SV;在CH中检测到3 893 609个SNP、360 528个InDel、12 756个CNV、20 501个SV。结果表明,2个番茄材料主要发生的变异类型为单核苷酸多态性(SNP)和小片段插入和缺失(InDel),与油菜、烟草和水稻的研究结果相吻合。对2个样本的KEGG通路进行比较,富集到的代谢通路包括吲哚生物碱生物合成、二萜生物合成、莽草酸途径生物碱的生物合成。为后续对变异基因分子标记的开发、分子标记辅助育种、基因克隆以及基因功能验证的研究奠定重要基础。
猜你喜欢
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
现代检验医学杂志(2015年6期)2015-02-06
实验动物与比较医学(2014年5期)2014-02-28
中国糖料(2013年1期)2013-01-22
医学综述(2011年18期)2011-12-10
