
基于SGCN 的化合物致癌性预测模型*
2022-07-22 06:32:42魏若冰何家峰邱晓芳
电子技术应用 2022年6期
魏若冰,何家峰,邱晓芳,刘 旗
(广东工业大学 信息工程学院,广东 广州 510006)
0 引言
由于技术的发展,新化合物的合成速度加快,每年诞生的化合物数以万计[1-2],传统的评价方法不可能对所有的化合物进行评估。并且近年来患癌人数不断增多[3],目前仍不清楚大多数的癌症是由于暴露于何种致癌化合物而导致的。世界卫生组织国际癌症机构(IARC)致癌清单中只有429 种化合物被归为具有致癌性物质,但仍有500 余种化合物未进行判定。传统的化合物致癌性评估主要通过实验测试进行,试验周期长且成本昂贵,不确定因素过多,因此迫切需要开发替代方法和工具来评估化合物的致癌性。
利用计算机进行毒性预测[4]是安全评价的重要手段,能够大幅度节省非临床安全评价试验成本,提高试验设计的科学性和准确性。随着机器学习的不断发展,支持向量机(SVM)、随机森林、神经网络(Random Forest)和K-最近邻(KNN)等机器学习算法已被广泛用于化合物毒性预测中[5-7]。此外,对致癌性化合物的预测也有一些报道。2004年,张晓昀等人[8]用人工神经网络中误差反向传播网络(BPNN)和径向基函数网络(RBFNN)对化合物的致癌性强弱进行了分类,模型的分类准确率达到了80%以上;2005年,张振山等人[9]用PCA 对分子描述符降维,利用决策森林的方法预测化合物致癌性;在2007年,谢莹等人[10]基于gSpan 算法,挖掘与已知毒性化合物具有相同字结构的化合物,进行未知化合物的毒性预测;2017年,梁倩倩等人[11]基于量化构效关系(QSAR)方法预测N-亚硝基化学物(NOCs)的致癌性,同年,阎爱侠等人[12]构建化合物的多维描述符,分别采用4 种机器学习方法(朴素贝叶斯、随机森林、多层感知机和支持向量机),模型的平均正确率达到74%±3%。
近年来,越来越多的研究人员把目光转向致癌化合物的研究,但是现有的模型评估化合物的致癌性能力有限。本研究从多个数据库整理了化合物致癌性数据,基于具有空间结构的原子特征建立了三维图卷积网络(Spatial Graph Convolutional Network,SGCN)。
1 数据和方法
1.1 数据收集
从世界卫生组织国际癌症机构(IARC)致癌清单和美国环境保护局(EPA)列出的安全化合物清单(SCIL)中收集数据。为了保证数据的准确性和可靠性,用以下标准来筛选和处理数据:(1)IARC 致癌清单中选择有足够证据证明对人类具有致癌性的化合物,剔除其他分类中对致癌证据有限和致癌证据不足的化合物;(2)SCIL 安全化合物清单中选择根据实验和建模数据,已被证实不具有致癌性的化合物;(3)从上述条件筛选的数据集中剔除无法确定分子结构的化合物。最终,获得了341 种实验数据,其中246 种致癌性数据为正样本,余下95 种不具有致癌性的数据为负样本,形成了最终的数据集。
1.2 数据集划分
从正负数据集中随机抽取数据:80%作为训练数据集(273 个分子)用于训练模型,10%作为验证数据集(34个分子)用于调整超参数,10%作为测试数据集(34 个分子)用于评估模型的性能。
1.3 分子编码
采用独立热(one-hot)对原子特征进行编码[13]。独热编码又称一位有效编码,其方法是使用N 位状态寄存器来对N 个状态进行编码,每个状态都有独立的寄存器位,并且在任意时候,其中只有一位有效,如图1 所示。同时,用RDKIT 计算原子和键的特征,包括原子的符号、原子连接的键的个数、原子的价态和键的类型、是不是共轭、在不在环中等。
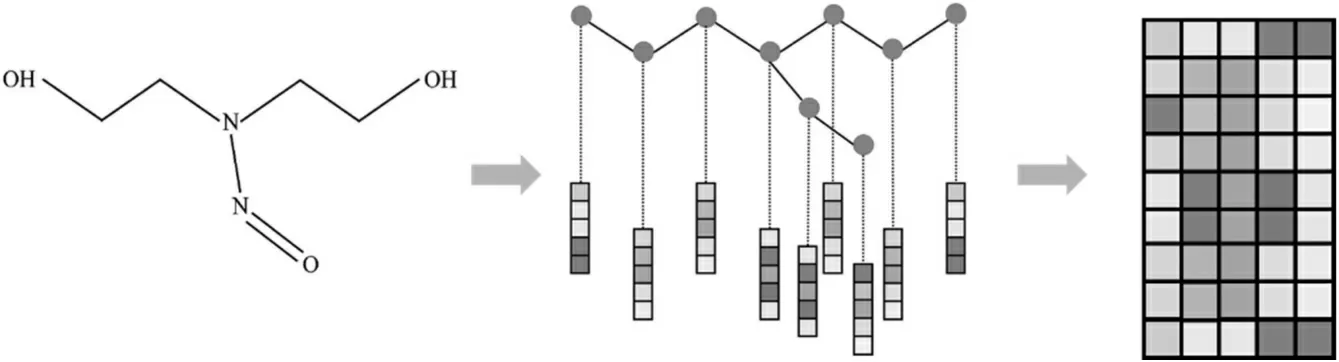
图1 对分子图进行独立热编码示意图
1.4 SGCN
本文中将分子的空间特性与传统的GCN 相结合,去预测分子的致癌性。大多GCN 模型使用二维分子图作为输入,通过特征矩阵和邻接矩阵去预测分子的性质[14]。然而,分子性质很大程度上受到空间中原子间相对位置影响,因此,在构建SGCN 模型时,把带有原子坐标的分子图也作为输入。
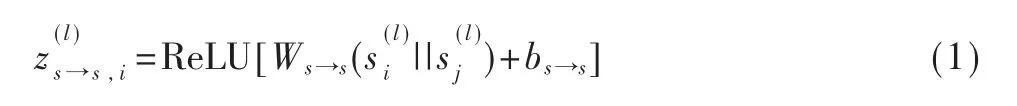
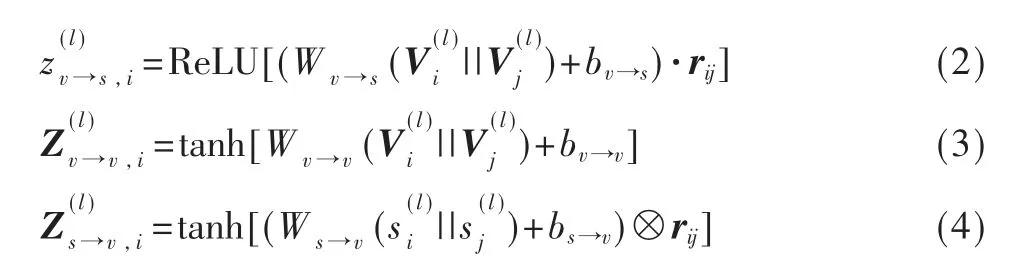
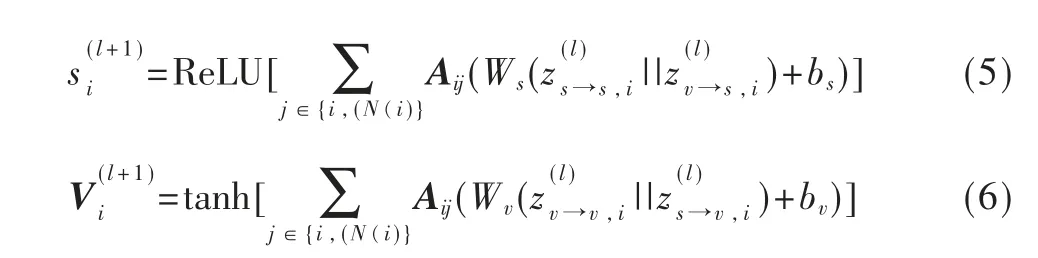
式中,A 是标准化后的邻接矩阵,W 和b 表示权重和偏置。空间GCN 由卷积层、特征构造层和全连接层3 个模块组成,如图2 所示。
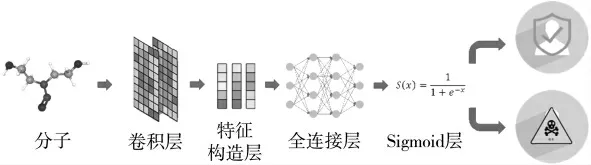
图2 空间图卷积流程图
在初始化特征时,节点的标量特征进行独立热编码形成60 个特征,而矢量特征被初始化为零。卷积层的第一阶段将每个节点的两个特征融合在一起,生成中间特征。在第二阶段,收集中间特征并沿着邻域进行汇总,从而产生更高级别的特征。通过卷积层,更新标量特征和矢量特征。经过卷积后,特征构造层通过两种策略收集节点上的特征:SGCNsum整合了节点上分布的所有原子特征,生成分子的标量和矢量特征;SGCNmax选取原子特征中取值最大的作为分子特征。生成的分子特征被送到具有ReLU 激活的全连接神经网络。最后,输出被扁平化处理后送到单层神经网络中来进行分类。
1.5 对比模型
对比模型包括GCN、多层感知机(Multilayer Perceptron,MLP)、随机森林(Random Forest,RF)、支持向量机(Support Vector Machines,SVM)、K-最近邻算法(K-Nearest Neighbors,KNN)、决策树(Decision Tree)、线性判别分析(Linear Discriminant Analysis,LDA)和XGBoost。GCN 模型由两个卷积层和一个全连接层构成,学习率为0.001。多层感知机中设置优化权重设置为adam,最大迭代300 次。余下机器学习模型从scikitlearn 库中调用,随机森林中建立子树的数量为20;支持向量机中核函数类型为径向核函数,布尔值为Truth;朴素贝叶斯分类器中拉普拉斯平滑系数设置为1,其余模型参数均设置为默认值。
1.6 模型评估方法
采用10 折交叉验证法来评估模型的预测性能和可靠性。在10 折交叉验证中,先将数据集划分为10 个大小相等的互斥子集,每个子集都尽可能保持了数据分布的一致性,之后,每次都用9 个子集作为训练集,余下的1 个子集作为验证集。然后,将交叉验证过程重复10 次。
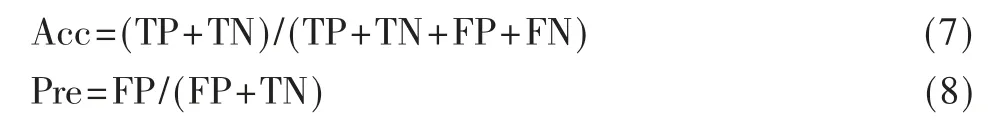
式中,TP 是真阳性,TN 是真阴性,FP 是假 阳性,FN 是假阴性。计算总体预测准确率(Acc)以对每个预测函数进行评估。此外,为了使评价更有效,加入了查准率(Pre)来进一步验证模型。查准率是计算模型判断为阳性的样本中有多少是真正的阳性。
2 结果和讨论
2.1 空间GCN 特征构造
在特征构造层以两种方式对特征进行构造,一种是将分布在节点上的所有原子特征相加(SGCNsum),形成新的矢量和标量特征;另一种是选取最大值的原子特征作为分子特征(SGCNmax),依据范数比对矢量特征的大小。根据表1 可以看出,SGCNmax和SGCNsum在对模型准确率预测在0.946~0.973 之间,查准率在0.939~0.951 之间,在GCN 为基础上准确率和查准率提高了约4.5%。在特征构造上,对比模型SGCNmax和SGCNsum在评估参数上的值,可以发现,SGCNsum除了在验证集的准确率略微低于SGCNmax,其余均高于SGCNmax,所以,在对分子致癌性进行预测时,特征构造中选取原子特征的最大值会使得模型效果偏好。
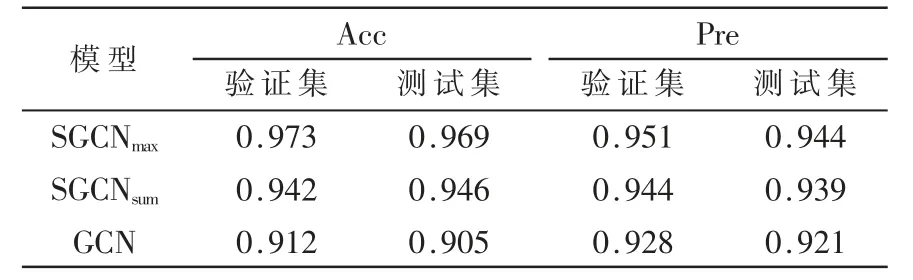
表1 模型的评估指标
2.2 对比实验
此外,还构建了7 个预测模型作为对比,7 个模型的整体准确率在0.810~0.861 之间,如表2 所示。
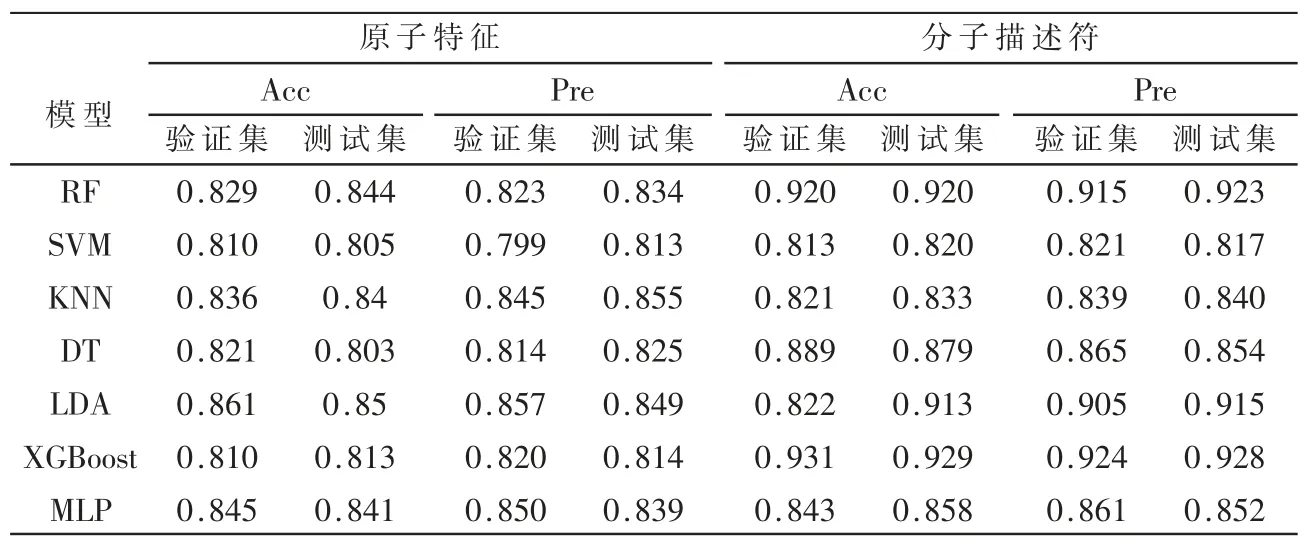
表2 基于原子特征和分子描述符的对比模型
在准确率评估中,表现最好的是RF 模型为0.844;在查准率评估中,表现最好的则是KNN 算法为0.855。在验证集中预测性能最好的LDA 算法在测试集中的表现同样优异,其总体预测准确率为0.861,查准率为0.849。除此之外,KNN 和RF 也表现出了较好的预测能力,KNN 在验证集的查准率达到了0.855。对比分析表2 中的模型可以看出,验证集和测试集中总体预测准确率和查准率基本相等,表明模型不存在过拟合的现象。将此表模型中表现最好的几个模型同SGCN 进行对比,可以看出SGCN 表现出了较为优异的性能。
2.3 提取分子描述符
在与7 种模型的对比实验中发现,与SGCN 和GCN进行对比时准确率差异过大达到了0.109,考虑到所有模型提取的特征为原子特征,SGCN 中的输入仅包括原子的特征矩阵还包括原子间的邻接矩阵和相对位置矩阵,而在对比实验中输入仅为原子特征,输入信息量相对较少且不全面,以用分子的信息代替原子的信息作为对比模型的输入。分子描述符[15]通过量化部分结构和物理化学性质来表达化合物的化学特征。使用函数调用rdkit生成数据集中所有分子的描述符,生成的描述符包含分子指纹、相对分子质量和部分电荷等200 维特征。将分子描述符作为输入用于7 种对比模型中,发现准确率有明显的上升,整个模型的准确率在0.821~0.931 之间,其中验证集中RF 和XGBoost 的准确率分别从0.829 和0.810 上升至了0.920 和0.931,除此之外DT 的准确率也上升了0.6,其他模型准确率没有变化或略微下降。
3 结论
本研究采用SGCN 模型对化合物进行了致癌性预测,可因此减少因条件限制而导致的化合物致癌性评估不足。此模型对273 种数据集和34 种外部验证数据集进行毒性分类,在34 种测试集中获得了96.9%的准确率和94.4%的查准率,表现出了评估化合物致癌性的优异性。通过进一步分析,发现用分子描述符作为特征时,RF 和XGBoost 模型效果准确率也达到90%以上,这两种模型同样也适用于化合物致癌性的分类。将SGCN 模型用于有毒气体分类上,准确率达到89%,说明此模型在化合物分类判定上也有一定的普适性。
该研究探索了基于原子空间特征结合SGCN 构建化合物致癌性分类模型的可行性,为化学物的健康风险评估提供依据,然而收集到的样本数和样本类别有限,需进一步增加样本量,使构建出的模型具有更好的泛化性和稳定性。
猜你喜欢
家庭医药(2022年9期)2022-05-30 10:48:04
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
食品安全导刊(2019年2期)2019-04-26 01:22:28
生命与灾害(2018年6期)2018-03-29 12:36:32
现代电子技术(2017年23期)2017-12-20 13:23:31
计算机应用(2016年10期)2017-05-12 11:02:20
计算机光盘软件与应用(2013年6期)2013-08-08 08:26:50
西安交通大学学报(2005年2期)2005-04-29 00:44:03
