
方解石和钾长石在模拟蚯蚓肠液中的初始溶解动力学机理及意义
2022-07-19 09:20:42蔡建林TENGHuiHenry王钺博张坚超
岩石矿物学杂志 2022年4期
蔡建林,TENG Hui Henry,王钺博,张坚超
(天津大学 表层地球系统科学研究院,地球系统科学学院,天津 300072)
蚯蚓活动对于土壤的重要性从达尔文时期就引起广泛关注(Satchell,2004)。蚯蚓主要通过摄食、排泄和钻洞等活动直接或间接地改变土壤生态系统的结构和功能,如形成土壤团聚体、增加土壤孔隙度等(Suzukietal.,2003)。和其他土壤动物相比,蚯蚓的肠道pH值接近中性,含氧量相对较低甚至无氧(Hornetal.,2003),这有利于厌氧微生物和兼性厌氧微生物的生长,与外界环境中的土壤微生物群落组成形成反差。土壤中的矿物被蚯蚓摄入后,经过大约12 h就被排出体外,单次摄入不足以使矿物和蚯蚓肠道中有机酸充分反应。经过蚯蚓长期反复地摄入和排出,土壤中的矿物才能得到充分溶蚀,进而改善土壤矿质组成,持续释放矿质元素。
随着蚯蚓的不断进食,其肠道内含有高浓度的可溶有机聚合物,这些聚合物经水解变为单糖。蚯蚓消化道后端糖的浓度低于前端,表明糖在肠道中被消耗(Wustetal.,2009a)。这些糖不仅可被蚯蚓消化吸收,也可在缺氧的肠道中被微生物所消耗,同时刺激产生乙醇和大量有机酸(Meieretal.,2018)。这些有机酸以小分子化合物为主,包括有甲酸、乙酸、丙酸、正丁酸、琥珀酸和乳酸(Wustetal.,2009a)。相比之下,土壤中这些化合物的浓度非常低甚至低于检测限,这表明这些有机酸在蚯蚓肠道中产生,而非来源于土壤(Wustetal.,2009b)。这些有机酸协同作用,在帮助蚯蚓消化营养物质的同时也有效地促进了摄入的土壤矿物的溶解。
有机酸对矿物溶解的促进作用及其反应机理一直备受关注,相关研究中涉及到的有机酸主要有甲酸、乙酸、柠檬酸、乳酸、琥珀酸等(Brantley and Stilligs,1996; Tengetal.,2006; 陈传平等,2008; Wustetal.,2009a; Yoshinoetal.,2010)。被溶解的岩石和矿物包括石灰岩、白云岩、方解石、钾长石、石英、含铁矿物、黏土矿物、毒砂等(Parsons,1994; 黄思静等,1995; Fredd and Fogler,1998; 向廷生等,2004; 陈传平等,2008; 王建萍等,2015; 朱文慧等,2015)。通过扫描电镜(SEM)、X射线衍射(XRD)、电感耦合等离子光谱仪(ICP)和离子色谱(IC)等表征手段(Agar and Geiger,2015; Yangetal.,2015; 邢俊涛等,2018; Zhangetal.,2019; Salimkhanietal.,2020),发现小分子有机酸能普遍促进矿物溶解。例如,草酸、柠檬酸、水杨酸、醋酸、邻苯二甲酸和邻苯二酚等均可以促进石英溶解(Bennett,1991),其中低浓度的草酸盐和柠檬酸盐溶液可使石英的溶解速率提高1倍以上(Blake and Walter,1999)。有机酸对长石同样具有明显的促溶作用,其中草酸和柠檬酸可使长石的溶解速率提升2~3倍(Blake and Walter,1999)。不同有机酸对石灰岩的溶蚀作用强弱依次为:乳酸﹥乙酸﹥柠檬酸﹥甲酸﹥草酸﹥丙酮酸(黄黎英等,2006)。在多数溶解实验中,有机酸浓度、溶液pH值和温度是影响矿物溶解的主要因素(黄思静等,1995; 罗孝俊等,2001; 郭春清等,2003; 向廷生等,2004)。与无机酸溶液相比,有机酸对矿物溶解的显著促进普遍被认为是有机酸根发挥了重要作用(Wogelius and Walther,1991; White and Brantley,1995; Drever and Stillings,1997)。有机酸促进硅酸盐矿物溶解、提高溶解度主要通过如下途径: ① 在矿物表面形成络合物,降低表面反应的活化能,提高矿物元素进入溶液的速率; ② 在流体溶液中,有机酸根与Si、Al结合形成络合离子,降低溶液中Si、Al的有效浓度(Pittman and Lewan,1994)。
前人的研究通常采取单一有机酸与矿物进行反应。在矿物溶解的研究中,一些复杂动力学模型,如表面络合模型、Monte Carlo模型和溶解步波模型(dissolution stepwave model)被用于对矿物的溶解进行模拟计算(Dyeretal.,2004; Kurganskayaetal.,2012; Martinetal.,2019),分析压力、温度、溶液pH值和有机酸配体等因素对矿物溶解的影响。而缩核模型和Hixson-Crowell模型更适合描述一些简单矿物的溶解过程(Lietal.,2015),尽管Hixson-Crowell模型通常用于药物缓释系统研究(Siepmann and Siepmann,2013)。但这些研究很少与土壤动物引起的矿物溶解联系在一起。蚯蚓对矿物溶解的体内原位研究因摄入时间短、反应后的矿物未发生明显变化且缺少溶液化学数据而难以开展。因此本研究模拟蚯蚓肠道,配置特定浓度和pH值的混合有机酸溶液,以土壤中常见的硅酸盐矿物(钾长石)和碳酸盐矿物(方解石)作为模型矿物进行溶解反应。通过体外模拟实验分析矿物溶解过程中溶液离子浓度的变化及反应后矿物表面形貌和粒度的变化,探究土壤矿物在蚯蚓体内的可能溶解过程及制约机制。
1 材料与方法
1.1 实验材料
本研究选择了土壤中常见的2种矿物,方解石和钾长石,代表了土壤矿物中成分(碳酸盐类与硅酸盐类)和结构(离子型与骨架型)的端员,溶解方式分别为化学计量溶解和非化学计量溶解。蚯蚓肠道混合有机酸采用优级纯的甲酸、乙酸、丙酸、丁酸、乳酸和琥珀酸行配置,浓度分别为3.26、18.33、8.11、13.64、5.93、4.44 mmol·L-1。为了模拟蚯蚓钙腺分泌的碳酸钙平衡肠道pH值,实验中使用碳酸盐调节混合有机酸pH值至蚯蚓肠道内呈中性。鉴于碳酸钙的使用会干扰溶出钙浓度的测量,因此采用碳酸铯(纯度≥99.9%)。
1.2 矿物溶解及表征方法
方解石和钾长石过200目筛,超纯水超声清洗15 min,60℃下干燥24 h备用。将4 g矿物粉末加入40 mL有机酸溶液或纯水中(7个一组)进行溶解实验,25℃下150 r/min振荡。每24 h每组中各取一份样品,测定pH值,离心(8 000 r/min,10 min)取上清液,其余样品更新40 mL初始溶液,实验持续7 d。上清液过0.45 μm滤膜,加入等体积3%二次蒸馏硝酸保存待分析。全部实验结束后,所有残余矿物固体60℃下干燥24 h待分析。
本研究利用ICP-OES(5110,美国Agilent公司)测定上清液中的离子浓度变化,并通过公式v=c/t计算离子的溶解速率,其中v表示溶解速率(mmol·L-1·h-1),t表示时间(h),c表示t时刻的离子浓度(mmol·L-1)。利用XRD(Mini Flex 600,日本Rigaku公司)分析固体的物相变化,扫描范围3°~70°,步长0.01°,扫描速度4°/min。利用SEM(Sigma 500,Ziess公司)观察反应前后矿物表面形貌变化。使用全自动比表面和孔径分析仪(Autosorb-iQ2-XR,美国Quantachrome公司)测量固体颗粒的比表面积随时间的变化,称取1.5 g样品于样品管中,110℃下脱气3 h,液氮-196℃状态下通过氮气吸附和解吸测量比表面积。利用超高粒度分析仪(MASTERSIZER 3000,英国Malvern公司)测量反应前后样品粒度分布变化。利用红外光谱(INVENIO,德国Bruker公司)分析残余矿物表面基团,称取1 mg样品与100 mg烘干的溴化钾(光谱纯,阿拉丁)在研钵中研磨,压片后上机测试。测试条件:分辨率4 cm-1,扫描次数16次,扫描范围4 000~400 cm-1。
2 结果
2.1 矿物溶解前后溶液pH值变化
矿物经有机酸和纯水中溶解24 h后,溶液pH值由中性变至弱碱性和碱性。经有机酸溶解后,体系pH值并未发生剧烈变化,方解石和钾长石的体系pH值变为弱碱性,分别在7.42~7.56和7.34~7.46间波动。有机酸溶解前后体系pH值变化不大的现象是由于有机弱酸的缓冲作用导致的,并且这种缓冲作用可以持续相当长的时间。溶解消耗的质子被部分电离的有机弱酸补充,在实验的7 d之内,溶液pH值均为较稳定的弱碱性。而在水溶液中,两种矿物的溶解导致体系pH值在第一天时变为强碱性(pH≈9.7),这是由于水溶液中没有弱酸的缓冲作用。而随着溶解的不断进行,方解石的体系pH值始终维持在9.68~9.89的强碱性范围,这是由于方解石的溶解在持续进行,溶液中的质子持续发挥作用。与此相比,钾长石经纯水溶解后,体系pH值不断趋向于弱碱性或中性,这表明钾长石的溶解速率在逐步减缓,溶液中质子的作用逐渐减缓(表1)。
由钙腺和肠道酸共同维持蚯蚓肠道环境的pH值稳定在近中性,本文中有机酸溶解矿物的模拟实验中,溶解前后体系pH值基本维持了中性至弱碱性的状态,模拟实验具可行性。
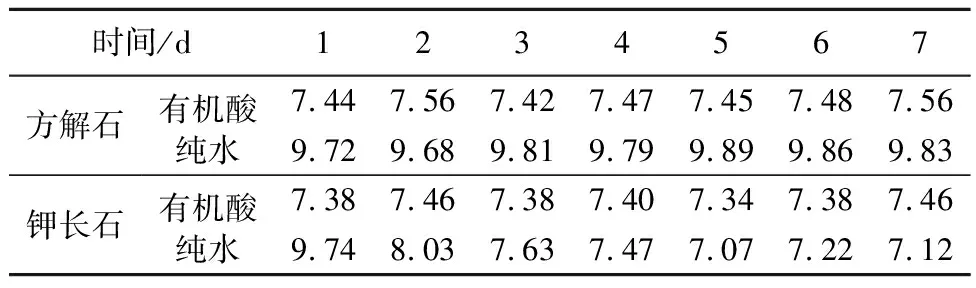
表1 有机酸和纯水溶解体系中pH值变化情况Table 1 pH value changes in organic acid and pure water systems
2.2 矿物溶解的离子浓度特征
方解石经第1天溶解后,有机酸溶液中的Ca2+浓度上升至2.61 mmol·L-1。之后第2至7天,Ca2+的累积溶出浓度与时间呈现出良好线性关系,以几乎稳定的溶解增量(~1.77 mmol·L-1)变化(图1a)。7天累积溶解浓度为13.2 mmol·L-1,平均溶解速率为7.86×10-2mmol·L-1·h-1。而在纯水中(图1b),Ca2+的累积溶出浓度与时间同样呈线性关系,Ca2+在第1天的溶出浓度为0.140 mmol·L-1,日增量为0.125~0.138 mmol·L-1,7天累积溶出浓度为0.937 mmol·L-1,平均溶解速率为5.58×10-3mmol·L-1·h-1。对比两者发现,尽管两种溶液的初始pH值相近,但方解石在有机酸溶液中的溶解速率比在纯水中高一个数量级。
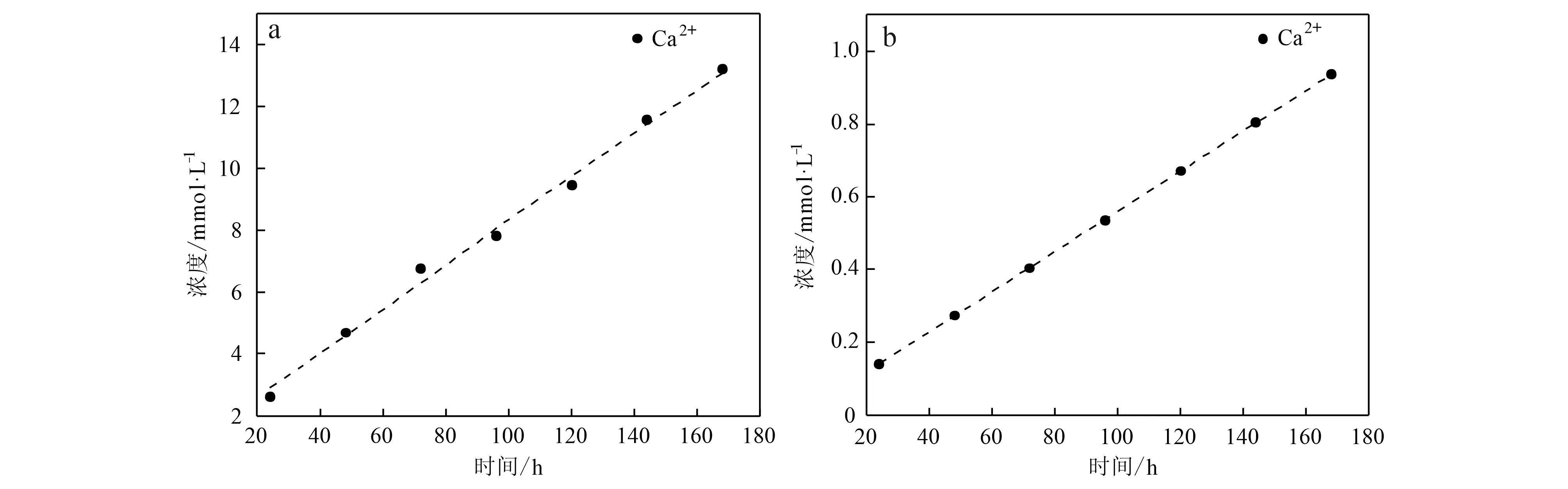
图1 方解石在有机酸溶液(a)和纯水(b)中溶出Ca2+的浓度随时间的变化Fig.1 Time dependent variations of Ca2+ concentration of calcite in organic acid (a) and pure water (b)
由于钾长石溶出的Al和Si水解形成溶胶,在过膜时被过滤掉,导致测试浓度低于实际浓度,因此钾长石的溶解仅表征K+的浓度变化。钾长石的累积溶解浓度与时间并不呈线性关系。钾长石在有机酸溶液中,第1天快速溶出1.08 mmol·L-1的K+。从第2天开始,K+的溶出浓度增量逐步降低,从0.337 mmol·L-1减小到0.011 5 mmol·L-1(图2a)。在溶解速率方面,反应初始时为最大值4.62×10-2mmol·L-1·h-1,随时间延长逐步降低,并且从第4天起接近于0(图2c)。钾长石在纯水中的溶解趋势与在有机酸中一致,即溶出速率逐步减小。溶出浓度增量从第1天的0.154 mmol·L-1减小到第7天的0.058 0 mmol·L-1(图2b)。第1天溶出速率为6.41×10-3mmol·L-1·h-1,第4天减小至2.49×10-3mmol·L-1·h-1并且稳定至第7天(图2d)。尽管第6天的溶解速率偏离拟合曲线(低于第5天和第7天),但在纵坐标的刻度范围内,其数值与前后相比未存在较大差异。对比两者发现,钾长石在有机酸溶液中的溶解速率比在纯水中高一个数量级,这与方解石在两种溶液中的溶解速率差异相同。
2.3 矿物及其溶解产物的晶体结构与表面形貌

在放大5 000倍的条件下,机械破碎的原始方解石颗粒边缘和棱角比较尖锐,表面平整光滑并附着少量碎屑(图4a); 方解石在有机酸溶液中溶解7天后,颗粒的边缘和棱角变得圆润,表面出现大小不等的呈四边形的溶蚀坑(图4b)。与图4b相比,经纯水溶解相同时间的方解石颗粒圆润度接近原矿,且溶蚀坑发育较弱(图4c)。同时,方解石的比表面积在有机酸中从1.243 m2·g-1降低至0.660 m2·g-1,而在纯水中降低至1.107 m2·g-1。
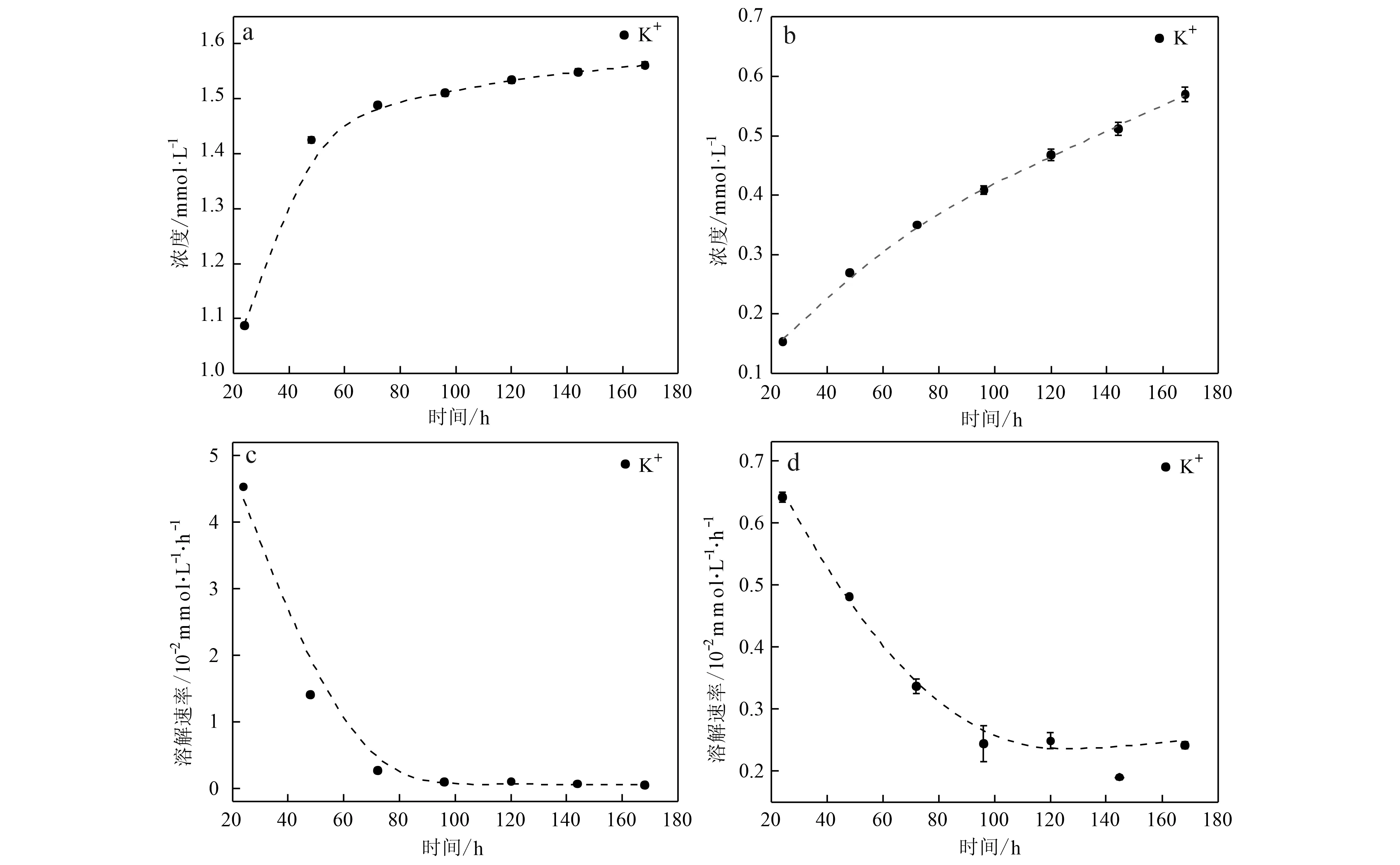
图2 钾长石在有机酸溶液(a, c)和纯水(b, d)中K+的浓度和溶解速率随时间的变化Fig.2 Time dependent variations of K+ concentration of K-feldspar and dissolution rate in organic acid (a, c) and pure water (b, d)

图3 方解石(a)和钾长石(b)溶解前后的XRD图Fig.3 XRD patterns of calcite (a) and K-feldspar (b) before and after dissolution
原始钾长石颗粒棱角和边缘同样尖锐,机械破碎痕迹和表面碎屑明显可见(图4d)。于有机酸和纯水中反应7天后溶解均较弱,颗粒表面仅见少量浅显溶蚀坑(图4e和图4f)。因此钾长石的比表面积始终接近2.581 m2·g-1。

图4 方解石和钾长石溶解前后的SEM图Fig.4 SEM images of calcite and K-feldspar before and after dissolution a—原始方解石; b—在有机酸溶液中溶解7天的方解石; c—在纯水中溶解7天的方解石; d—原始钾长石; e—在有机酸中溶解7天的钾长石; f—在纯水中溶解7天的钾长石a—the raw calcite; b—the calcite dissolved in organic acid for 7 days; c—the calcite dissolved in pure water for 7 days; d—the raw K-feldspar;e—the K-feldspar dissolved in organic acid for 7 days; f—the K-feldspar dissolved in pure water for 7 days
2.4 矿物及其溶解产物的粒度
方解石经有机酸溶液溶解后,粒度分布向右偏移,表明颗粒粒度逐渐增大(图5a),这可能是由于小颗粒溶解而大颗粒富集。相比之下,在纯水中第1天到第4天峰位向右偏移,颗粒粒度逐渐变大,但第4天到第7天较小粒径的颗粒占比增加,这可能是由于大颗粒破碎分解成更小颗粒(图5b)。
钾长石在有机酸中的主要粒度未表现出显著变化,但较小粒度部分逐渐减少(图5c)。在纯水中,无论粒度大小,粒度分布在整个实验期间保持稳定,没有明显的变化(图5d)。
2.5 矿物及其溶解产物的表面化学特性
钾长石经有机酸溶解的红外吸收峰位未发生漂移(图6a),随着时间的增长,3 460 cm-1处吸附水和表面羟基的伸缩振动峰强度增加,同时,1 008 cm-1处Si(Al)—O伸缩振动峰强度降低(表2)。经纯水溶解的红外图谱呈现出与有机酸溶液中一致的规律,即随着时间的增长,吸附水和表面羟基的伸缩振动峰强度同样有所增加,Si(Al)—O伸缩振动峰强度同样降低(图6b)。但对比两者发现,钾长石的Si(Al)—O伸缩振动峰强度在经过有机酸溶解后降低得更明显。方解石是一种离子化合物,化学计量溶解,在溶解表面通常不产生二次相。因此,本研究中未对方解石进行红外表征。
3 讨论
3.1 配体对矿物表面溶解的影响(配体效应)
有机酸(草酸、柠檬酸、琥珀酸、甲酸、乙酸、丙酸以及氨基酸等)与各种矿物(硅酸盐、碳酸盐、磷酸盐)和岩石(花岗岩)的相互作用已被广泛研究(表3)。矿物和岩石的溶解速率、表面形貌和物相变化等方面的表征结果均表明有机酸对矿物和岩石的溶解具有促进作用。

图5 方解石和钾长石在有机酸溶液(a、c)和纯水(b、d)中的粒度分布随时间的变化Fig.5 Time dependent variations of particle size distribution of the calcite and K-feldspar dissolved in organic acid (a, c) and pure water (b, d)

(1)

(2)
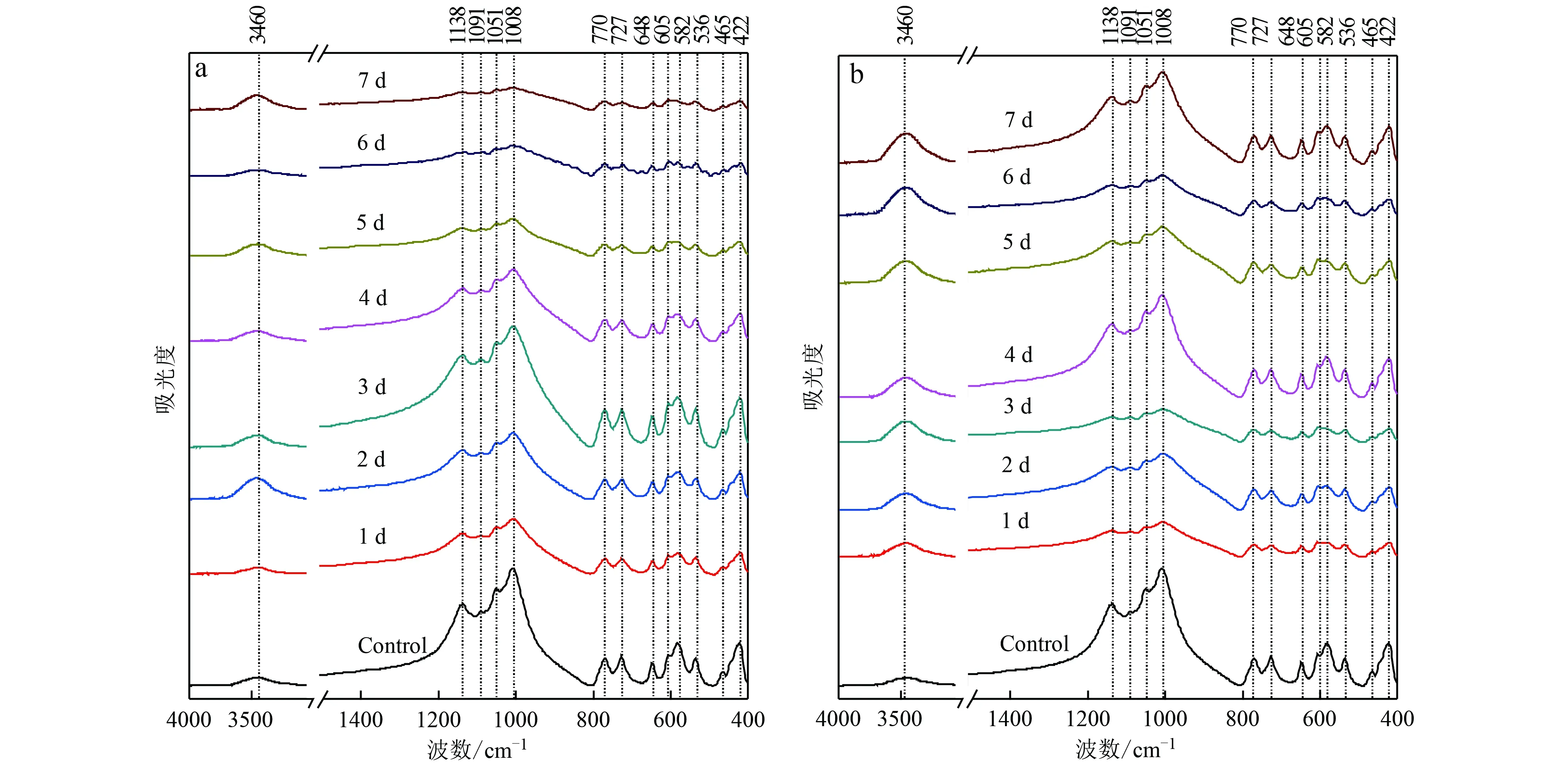
图6 有机酸(a)和纯水(b)中溶解前后钾长石的FTIR图谱Fig.6 FTIR spectra of K-feldspar dissolved in organic acid (a) and pure water (b)
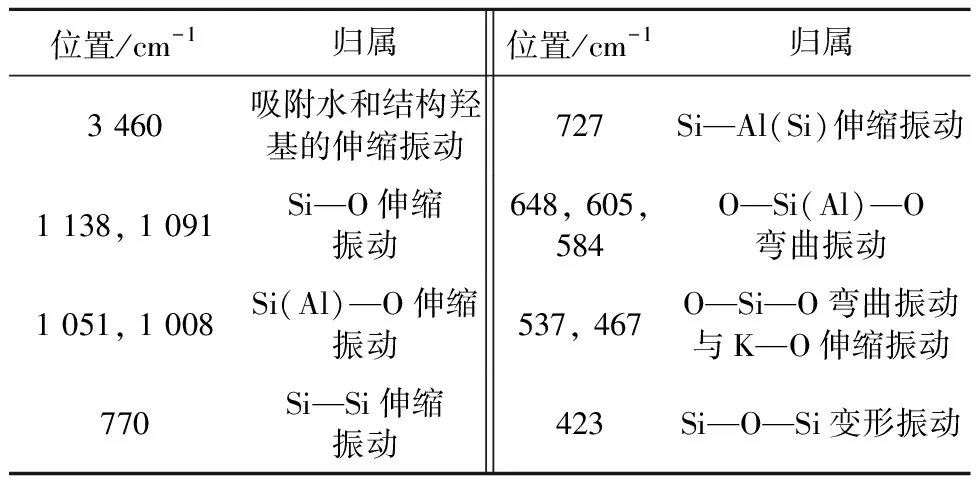
表2 钾长石FTIR振动峰的归属Table 2 The attribution of FTIR vibration peaks of K-feldspar
有机酸(草酸、柠檬酸、乳酸和琥珀酸)对方解石溶解的促进作用已有报道(王建萍等,2015),其作用机理为在溶液中和溶解表面形成有机钙络合物,琥珀酸、柠檬酸、 乳酸和草酸均具有多官能团,能够与溶液中Ca2+络合形成多元络合物,与Ca2+在溶液中的络合常数(logK)分别为2、4.9、1.62和3,形成络合物的强度较大。因此,有机配体降低了溶液中Ca2+浓度,进而促进溶解。Teng等(1997,2006)研究了天冬氨酸和琥珀酸与方解石的相互作用,并记录了有机分子与表面位点结晶方向的特异性结合,结果表明,表面络合物的形成会降低方解石晶格中钙与氧的结合键能,从而促进钙的分离。
配体效应同样适用于有机酸对长石的促进溶解。相同pH值的有机酸比无机酸对长石的溶解能力更强(安波等,2018)。多种小分子有机酸(酒石酸、柠檬酸和草酸等)均能显著促进含钾矿物(云母、长石)中K+的释放,与纯水相比促进效果可达到1.5~2.0倍(王瑾等,2013)。在模拟典型成岩环境的条件下,乙酸盐溶液和乙酸盐+草酸盐溶液均能显著增加钠长石的溶解度,与蒸馏水相比,其溶解度分别提高了2倍和3.4倍(Franklinetal.,1994)。Blake等(1996)研究了在80℃且pH值为6的情况下,几种有机酸分别对正长石的溶解情况,结果显示正长石在10 mM的草酸和柠檬酸溶液溶解过程中Si和Al的浓度是乙酸缓冲溶液的近3倍,这表明在相同浓度下,柠檬酸和草酸对长石的溶解能力均比乙酸强。此外,有机酸还可以促进花岗岩的溶解,花岗岩在pH=6的有机配体溶液中,所有元素的释放都显著增加,促进作用依次为柠檬酸盐>草酸盐≈丙二酸盐(Neamanetal.,2006)。
与方解石的化学计量溶解不同,长石在中性和偏酸性条件下的溶解方式是非化学计量溶解(Stober and Bucher,2007)。长石中的碱金属与碱土金属阳离子通过离子键与硅氧骨架相连。在溶解初期,溶液中的H+与长石表面的K+、Na+、Ca2+等阳离子发生离子交换反应,使碱性阳离子优先被溶出进入溶液 (薛伟伟等,2015),但矿物颗粒的粒度基本保持不变
表3 小分子有机酸对矿物的溶解
Table 3 Minerals dissolution in small molecular organic acids
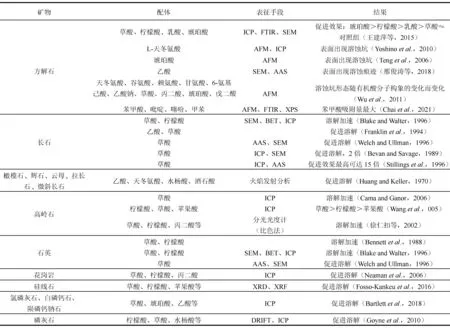
(图5c、5d)。研究发现这些金属离子的溶出浓度随着时间表现出前期缓慢上升,之后保持不变的趋势,例如,长石在pH = 6.25的乙酸溶液中溶解,溶液中K+的浓度从大约2 mmol·L-1经过400 h上升至大约4 mmol·L-1,之后当反应时间达到1 000 h时K+浓度依旧保持稳定(方晓玲等,2019)。在长期溶解实验中,硅氧骨架表面的Si—O和Al—O键经过水合和水解作用,最终导致了骨架的解体,这与本研究中钾长石表面的(Si)Ai—O振动信号减弱和表面羟基伸缩振动信号增强的实验数据(图6,表2)相一致。此外,长石骨架的破坏同样是非化学计量的,在长期溶解实验中,表面的Si逐渐富集,如长石溶解过程中形成的硅膜(Si-coating)是Al优先释放的结果(Crovisieretal.,1983)。但另外有研究表明,硅膜更有可能是通过溶液相聚合形成(O’Neil and Taylor,1967)。但无论是哪种情况,这种硅膜不会在本实验所处的初始溶解期间形成。
在有机酸溶液中,络合物的络合常数通常随着官能团(结合位点)的数目增加而变大(表4)。本研究采用的有机酸为甲酸、乙酸、丙酸、丁酸、乳酸和琥珀酸配制的混合有机酸溶液。虽然表面复合物的络合常数就像§-CaCO3-L-(方程式2)的络合常数是未知的,但是表面络合模型表明,表面络合常数的相对大小与溶液络合常数具有同样的趋势(Stumm,1987)。本研究应用的有机酸中,琥珀酸和乳酸比其他有机酸络合效果更强。这是因为甲酸、乙酸等均为一元羧酸,分子结构中仅含有1个—COOH,而像琥珀酸属于二元羧酸,分子中有2个—COOH,乳酸虽然属于一元羧酸,但其分子中包含的—OH同样为络合提供了结合位点。因此琥珀酸和乳酸作用的产物更稳定,促使反应朝有利于溶蚀的方向进行。据此,可以推断出这些有机酸对方解石溶解的贡献的顺序为琥珀酸、乳酸和丙酸>甲酸、乙酸和丁酸(表4)。同样的推断也适用于钾长石的溶解,这与K+从表面分离有关,K+与有机配体形成表面络合物或溶液络合物,因此推断K+与甲酸、乙酸、丙酸、丁酸、乳酸和琥珀酸之间的反应中乙酸和具有多官能团的琥珀酸发挥主要作用。
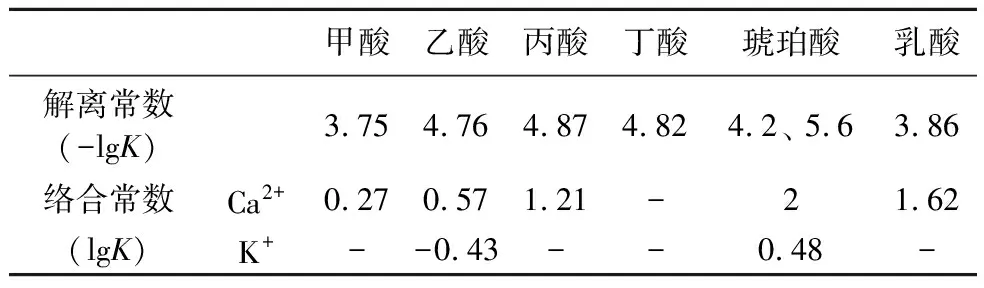
表4 常见有机酸的解离常数及与Ca2+和K+在溶液中的络合稳定常数(25℃) Table 4 Dissociation constants of common organic acids and their complexation stability constants with Ca2+ and K+(25℃)
3.2 溶解动力学模型
两种矿物的化学溶解数据(图1,图2)和粒度变化数据(图5)表明,方解石和钾长石通过不同的化学途径溶解,符合已知的化学计量溶解与非化学计量溶解机制(Shotyk and Nesbitt,1992; Kaufmann and Dreybrodt,2007)。由于非化学计量溶解,钾长石在短期实验中粒度(或表面积)的变化可以忽略不计。因此,其溶解动力学可以用初始速率法来解释。而方解石的溶解动力学不能仅通过溶液化学来解释,因为颗粒粒度(或表面积)的变化不容忽视。因此,将矿物粒度与溶解数据联系起来就需要一条更好的途径,但矿物的粒度分布与其溶解速率之间的关系通常比较复杂,通常这种关系并不是线性相关(根据Gibbs-Thompson效应小颗粒溶解得更快)。粒度分布数据(图5,颗粒粒度逐渐变大)表明,小颗粒完全溶解的速度比大颗粒的溶解变小的速度快。因此,本文中尝试运用动力学模型[缩核模型(shrink core model)和Hixson-Crowell模型]从不同的角度对方解石的溶解进行探讨。
(1) 初始速率法
在钾长石溶解初期,碱金属在溶解过程中通过离子交换反应从晶格中释放出来。在多数实验中,这种交换机制可逆,并影响最外层2或3个晶格单位。如图7所示,钾长石的晶体结构可以被看作是由平行于x轴的一系列Si(Al)—O四面体的之字形链对组成。其中,每一对双链之间构成四元环,相邻层四元环之间形成间隙,K+或Na+作为间隙离子存在于开放空间中。溶液中的H+与矿物表面的间隙离子发生交换反应,这一步反应相对快速,表面Si(Al) —O框架破坏之后才能进一步交换内部的间隙离子。在本研究中,反应时间控制在7天内,因此认为钾长石的溶解处于第一步,即H+与钾长石表面的间隙离子发生交换反应。
从图2可以看出,K+在有机酸溶液和纯水中的溶解速率呈现出不同的规律。为定量描述这种变化,使用方程对其进行拟合。如图8a所示,在有机酸溶液中,前3天溶解速率呈指数降低,符合方程:
R=kt-2(1)
其中,R表示溶解速率(mmol·L-1·h-1),k为常数(mmol·L-1·h)。拟合后k=2.63×103(mmol·L-1·h)。从第3天到第7天,溶解速率与时间呈现出线性关系,拟合斜率为-1.91×10-3。相较于有机酸溶液,K+在纯水中的溶解速率变化更趋近于线性关系,拟合斜率为-2.81×10-3。当有机酸溶液中的溶解速率与时间呈现出线性关系时,其溶解速率在数值上低于纯水中。由于本研究实验时间较短,可以认为K+的溶解主要发生在表面,钾长石的Si(Al)—O骨架结构的变化可以忽略不计。
图8a表明了钾长石在有机酸溶液和纯水中溶解速率随时间的变化,但未揭示在初始时刻(t=0)的溶解速率。为进一步探究本系统的溶解过程,本文采用了初始速率法拟合溶液中K+浓度变化过程,并计算得出初始溶解率(Lasaga,1998)。初始速率法假设浓度变化符合多项表达式
[K+]=a+bt+ctn(2)
其中,a、b和c为拟合参数,n为多项式的指数且n>1,t=0时d[K+]/dt的结果可以确定为初始溶解速率,在数值上与b相等。钾长石中K+的溶解浓度变化的拟合如图8b所示。根据拟合结果,K+在有机酸的溶出过程在前3天拟合为非线性方程
[K+]=a+bt+ct1.1(3)
其中,d[K+]/dt=b+1.1ct0.1,当t=0时,d[K+]/dt=b=0.258 mmol·L-1·h-1,即钾长石在有机酸溶液中的初始溶解速率为0.258 mmol·L-1·h-1。而从第3天到第7天,K+在有机酸溶液中的浓度变化拟合为线性方程
[K+]=d+et(4)
其中,e=7.66×10-4mmol·L-1·h-1,这表明从第3天开始,K+的溶解速率急剧下降。相比之下,钾长石中K+在纯水中的溶解过程拟合为非线性方程:
[K+]=a+bt+ct1.1(3)
其中,d[K+]/dt=b+ 1.1ct0.1,当t=0时,d[K+]/dt=b=0.022 mmol·L-1·h-1,即钾长石在纯水中的初始溶解速率为0.022 mmol·L-1·h-1,比有机酸溶液中的初始溶解速率低一个数量级。

图7 钾长石晶体结构示意图(黄色形状表示[SiO4]或者[AlO4]四面体,红色球代表氧原子,蓝色球代表钾离子)Fig.7 The crystalline structure of K-feldspar (the yellow tetrahedrons represent [SiO4] or [AlO4] tetrahedrons, the red spheres represent oxygen atoms, the blue spheres represent the potassium cations)

图8 钾长石中K+的溶解速率(a)及溶解浓度(b)随时间变化与方程(1、3、 4)的拟合结果Fig.8 Time dependent variations of dissolution rate (a) and concentration (b) of K+ in K-feldspar and the fitting curves according to equation (1, 3, 4)
图8a中溶解速率的快速下降可以解释为K+释放仅限于钾长石的表面,当表面的K+释放完毕后,内部的K+与H+继续发生交换反应,因需要更多能量而溶解速率降低。因此在有机酸中的前期溶解速率较高,溶出浓度随时间变化符合非线性方程,当表面K+充分溶出后溶解速率迅速下降,矿物内部的K+以较低的速率缓慢溶出且溶出浓度随时间变化符合线性方程。相比之下,在纯水中,由于K+溶解速率较低,钾长石的表面K+未完全溶出而浓度随时间变化全程符合非线性方程(图8b)。同时,初始溶解速率通过拟合计算得出,分别为0.258 mmol·L-1·h-1和0.022 mmol·L-1·h-1,这表明有机酸根促进了K+的释放,一方面有机酸能够提供更多的H+,提升了K+的溶解速率; 另一方面有机酸根还可与K+形成络合物,促进了K+进一步的溶解。这与溶解长石结构变化不大的假设一致。
(2) 缩核模型(shrink core model)
缩核模型最早由Yagi和Kunii(1955)提出,并在Octave Levenspiel的化学反应工程教材中加以完整描述(Wantaetal.,2016)。该模型假设,溶解过程中主要的化学反应发生于尚未反应的颗粒核心和生成物的相界表面,而溶解是由球型颗粒表面自外向内各向同性进行的,各溶解阶段中未反应的颗粒核心总保持着相同的球形。式5~7分别描述了反应速率受产物外扩散(k1)、溶液中反应物内扩散(k2)和表面化学反应速率(k3)控制时的动力学规律(Bidari and Aghazadeh,2015; 王新宇等,2017; 缪彦等,2019):
x=k1t(5)
1-3(1-x)0.67+2(1-x)=k2t(6)
1-(1-x)0.33=k3t(7)
式中,x为方解石的Ca溶出的质量分数(%);k1、k2、k3为反应控制模型动力学常数;t为时间(h)。将图1中Ca2+的溶解数据分别代入3个方程进行拟合,结果如图9所示,可以看出方解石的溶解数据与缩核模型的拟合结果较好。根据图9,方解石在有机酸溶液中和纯水中的反应控制动力学常数如表5所示。在有机酸和纯水中,3个溶解速率常数大小呈现出一致的规律,即k1>k3>k2。3个溶解速率常数在有机酸溶液和纯水中分别相差14.526倍、19.231倍和13.005倍,相对大小变化比较接近。这表明方解石在产物外扩散(k1)、溶液中反应物内扩散(k2)和表面化学反应(k3)的共同作用下发生溶解反应,其中溶液中反应物内扩散(k2)即溶液中质子或配体扩散到达方解石反应表面的速率是限制步骤。

图9 方解石在(a, c, e, g)有机酸溶液和(b, d, f, h)纯水中溶解随时间的变化与方程(5, 6, 7, 8)的拟合结果Fig.9 Time dependent variations of dissolution of calcite in organic acid(a, c, e, g) and pure water (b, d, f, h) and the fitting curves according to equation (5, 6, 7, 8)

表5 方解石拟合缩核模型的反应控制动力学常数Table 5 Reaction-control kinetic constants of calcite fitted by shrinking core model
(3) Hixson-Crowell模型
当方解石颗粒溶解时,其质量随时间而减小,这可用Hixson-Crowell立方根溶解动力学方程来描述。
W0为初始的矿物质量(mg),W表示剩余的矿物质量(mg),K为立方根溶解速率常数(mg1/3·h-1)。此方程基于下列假设: ① 溶解作用发生于矿物固体颗粒的表面; ② 固体颗粒表面的周围不断受到均匀搅拌; ③ 在整个溶解过程中,固体颗粒的几何形状接近球形且始终不变。在表6中分别列出了方解石在有机酸溶液和纯水中的溶解速率常数K。从表中数据可以得出,与纯水中相比,有机酸中的溶解速率常数随时间增长略降低。方解石在有机酸中溶解速率常数K1的平均值为4.69×10-4mg1/3·h-1,在纯水中的溶解速率常数K2的平均值为2.99×10-5mg1/3·h-1,二者相差一个数量级。

表6 方解石在有机酸溶液和纯水中的溶解Table 6 Dissolution of calcite in organic acid and pure water
方解石在有机酸溶液中和纯水中溶解的方程模拟如图9所示,可以看出方解石的溶解过程基本符合Hixson-Crowell方程。根据模拟结果,方解石在有机酸中的溶解速率常数K1’ = 4.31×10-4± 1.19×10-5mg1/3·h-1; 在纯水中的溶解速率常数K2’ = 2.93×10-5±5.55×10-7mg1/3·h-1,二者相差一个数量级。结合方解石溶解过程中粒度的变化(图5a、5b),可认为溶解过程中的粒度变化不会影响Hixson-Crowell方程中溶解速率常数,这与方解石在有机酸和纯水中的溶解速率的数据结果相一致(图1)。
4 结论
在有机酸溶液中,方解石溶解的残余固体表面出现了数量多、程度深的溶蚀坑,溶解时较大颗粒逐渐富集,其结晶度上升。钾长石溶解的残余固体表面仅有少量浅显溶蚀坑,溶解时整体颗粒粒度基本未发生变化,但其结晶度下降,且固体表面吸附水和结构羟基的吸收峰逐渐增强。以方解石的溶出率为变量的缩核模型的计算结果表明方解石溶解主要受到反应物内扩散的影响,以溶解质量为变量的Hixson-Crowell方程同样定量描述了方解石的溶解过程。在相同初始pH值的体系中,有机酸溶液的溶解速率比在纯水中高一个数量级。有机酸溶液中钾长石表面的K+迅速溶出,而内部K+则以较低的速率溶出,在纯水中表面K+持续以较低速率缓慢溶出,二者同样相差一个数量级。
本研究通过体外模拟实验揭示了矿物在模拟蚯蚓肠道有机酸的溶液中加速溶解,其作用机制可以通过溶液化学和表面络合反应加以解释,动力学分析量化了溶解过程。但需要注意的是,本研究中模拟的蚯蚓肠道化学结果需经过进一步分析才能用于描述生态系统中蚯蚓对矿物的风化。
猜你喜欢
硅酸盐通报(2022年8期)2022-09-08 04:25:42
中国化肥信息(2021年2期)2021-07-23 01:49:00
矿产综合利用(2021年3期)2021-07-14 03:46:54
矿产综合利用(2020年1期)2020-07-24 08:50:46
西南石油大学学报(自然科学版)(2019年4期)2019-11-04 00:34:28
北方人(2018年16期)2018-08-20 06:01:22
爱你(2016年4期)2016-12-06 05:15:27
中国农资(2016年1期)2016-12-01 05:21:15
照明工程学报(2016年3期)2016-06-01 12:18:01
中国资源综合利用(2016年5期)2016-02-03 02:56:16
