
氧化物负载的单原子催化剂研究进展
2022-07-18 03:56刘小刚陈文婕
信阳师范学院学报(自然科学版) 2022年3期
刘小刚,张 欣,陈文婕
(信阳师范学院 化学化工学院, 河南 信阳 464000)
0 引言
多相催化反应通常发生在固体催化剂的表界面,体相原子在整个反应过程中的作用微不足道,即使将催化剂的颗粒尺寸减小到纳米级,表面原子的暴露比例也很低。贵金属催化剂(如Pt、Pd和Ru)已被广泛应用于能源与环境催化领域,将贵金属颗粒尺寸减小到原子级别,并均匀分散在氧化物载体材料上,不仅可以降低贵金属的使用量,而且可以最大化地发挥其催化效率。2011年,中国科学院大连化学物理研究所张涛团队成功制备了Pt1/FeOx单原子催化剂,由此提出单原子催化剂(Single-Atom Catalysts,简称SACs)的概念[1]。
本文以金属-氧化物之间的相互作用为出发点,简要介绍氧化铁(FeOx)、二氧化钛(TiO2)、氧化铜(CuO)、氧化锌(ZnO)和二氧化硅(SiO2)等典型氧化物负载SACs的合成方法、表征手段以及在催化反应中的应用(见图1)。
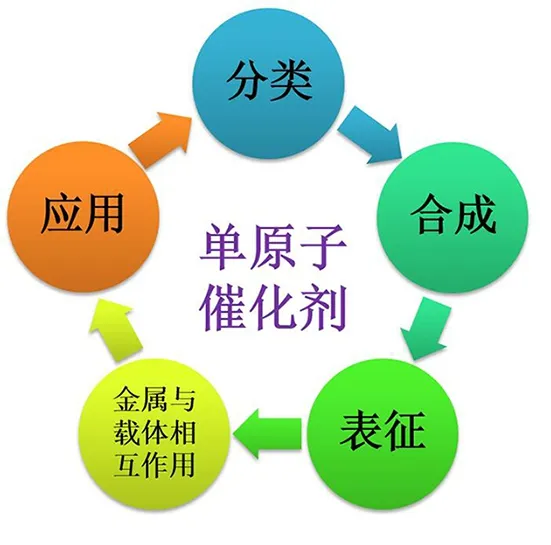
图1 SACs的分类、合成、表征、金属-载体相互作用及应用概述图Fig. 1 Schematic overview of the classification,preparation,characterizations,interaction between supports and metal and applications of SACs
1 不同类型的金属-氧化物单原子催化剂
氧化物具有独特及可调变的物化性质、丰富的表面缺陷位点以及—OH基团,可以作为单个金属原子的锚定位点,因此是单原子金属理想的载体材料。科学家对Fe3O4负载的单原子催化剂的研究表明,Fe3O4(001)界面经过表面重构,不仅可以容纳孤立的Pd、Pt或Au原子,而且容易形成Fe空位。QU等[2]报道了在Mo1/Fe2O3表面存在一个扭曲的八面体结构,其中Fe原子被Mo原子取代。其他金属(如Pd[3]和Ir[4])也可以沉积在FeOx表面,生成高度分散的多相催化剂。除空位外,孤立的金属原子也可以通过金属和无缺陷氧化铁之间的特殊相互作用来稳定。QIAO等[1]在700 ℃以上的氧气气氛下煅烧Pt NPs,制备了热稳定的Pt1/Fe2O3SACs。尽管Fe2O3载体缺少足够缺陷位点,移动的Pt原子也能被原位形成的共价Pt—O—Fe键所稳定。
TiO2是一种价格低廉、无毒且化学性质稳定的金属氧化物,在自然界中通常存在金红石、锐钛矿和板钛矿三种晶相,其中金红石和锐钛矿相TiO2已被广泛用作催化剂载体材料[5-8]。例如,V[9]、Cr[10]和Fe[11]成功地沉积在金红石/锐钛矿相TiO2上,形成各种单原子催化剂。近年来,板钛矿相TiO2纳米棒可以作为优异的单原子催化剂载体,将一元或多元过渡金属锚定在板钛矿 TiO2纳米棒的表面[12]。LIU等[13]制备了超薄TiO2纳米片负载的Pd催化剂,Pd负载量高达1.5%。另外,负载在TiO2纳米材料上的孤立金属单原子催化剂在热/电催化和化学传感器领域具有较好的应用。例如,Fe掺杂TiO2大大提高了N2电催化还原的稳定性。TiO2负载的Pd SACs、Pt SACs在酮和醛的氢化反应中表现出优异的化学活性和选择性[14]。
除 FeOx和 TiO2外,NiO 也是SACs 常用的载体材料之一。WANG等[15]将NiO在H2IrCl6前驱体溶液中浸渍、空气中退火,成功合成了超高负载量(质量分数为18%)的Ir/NiO单原子催化剂。X射线光电子能谱(XPS)和X射线吸收近边结构光谱(XANES)表征结果表明,Ir/NiO SACs中Ir的化合价约为+4,接近于本体IrO2中Ir的化合价。
将单分散的金属阳离子还原成金属态会削弱金属-氧化物的相互作用,形成金属簇和纳米粒子。然而,当 CuO和ZnO作为载体时,它们能够稳定处于金属状态的孤立金属原子。例如,单层CuO薄膜以表面缺陷作为势阱,通过双齿O—Au—O螯合来捕获Au原子[16]。在CO氧化过程中,CuO晶格中的氧空位只在活性中心的外围形成。当载体从CuO变成Cu2O时,单个 Pt 原子以中性电荷状态存在,这在氧化物担载的SACs中鲜有报道[17]。LANG等[18]制备了ZnO纳米线负载的Rh SACs,由于Rh和ZnO的强相互作用,Rh保持单一分散状态且为金属态。在苯乙烯的加氢甲酰化反应中,负载在ZnO上的Rh SACs具有优异的化学选择性[18]。
SiO2被认为是固定孤立金属原子不可还原的氧化物载体。水合金属铝硅酸盐化合物称为沸石,过渡金属物质很容易负载在沸石中,使用沸石作为载体可以提高SACs的反应活性和选择性。TANG等[19]报道了包封在ZSM-5孔中的Rh1O5催化剂,在温和条件下具有较高催化甲烷生成乙酸的转换效率,而Rh SACs负载在无孔氧化物Al2O3和SiO2上的活性明显较低。
2 氧化物负载SACs的合成方法
制备氧化物负载的单原子催化剂是研究其结构和催化行为的前提。虽然金属前驱体可以较容易地转移到催化剂的载体表面,但是保持金属原子在溶液中或载体上的单一分散仍具有挑战性。根据不同的金属前驱体,氧化物负载单原子催化剂的合成路线通常可分为 “自下而上”和“自上而下”两种。单核金属前驱体一般选择“自下而上”策略,将金属直接沉积在载体表面或与载体前驱体材料混合。而金属胶体/纳米粒子/团簇或金属块体一般采用“自上而下”的策略来制备SACs。一般而言, 大多数SACs的制备方法集中在湿化学方法,包括共沉淀、浸渍、沉积-沉淀和光/电化学沉积等。
共沉淀法是合成金属氧化物负载SACs最可靠的方法之一,几乎所有的金属氧化物SACs都可以通过金属盐与碱溶液的共沉淀来制备。成核过程中,可溶的单金属物种与氧化物载体紧密混合,使得催化剂中的单原子均匀分布。例如, QIAO等[1]将H2PtCl6和Fe(NO3)3溶液混合,滴加到Na2CO3水溶液中,随后活化处理制备了Pt1/FeOxSACs。然而,这种方法的缺点是会浪费一些嵌入到大块载体中的活性金属物种。
浸渍法是合成单原子活性金属位点暴露在氧化物载体表面的一种方法,几乎可以制备任何氧化物担载的SACs。该方法操作简单,将载体浸入含有单一金属前驱体的溶液中,随后进行适当的活化处理,包括干燥、煅烧和还原等方法。然而,金属物种通常因被锚定到载体的缺陷位和台阶而随机分散在载体表面。例如,CeO2和TiO2可以产生更多的空位缺陷来捕获单个金属原子,并改变金属原子与载体之间的相互作用[20-21]。
沉积-沉淀方法合成高负载单原子催化剂,首先要对氧化物材料表面微纳结构进行适当修饰和改性,如:创造更多的锚定位点和增大载体的比表面积等。其次将载体分散在金属盐/络合物前驱体溶液中,通过调节溶液的pH值,金属盐进一步沉淀为金属氢氧化物或碳酸盐。在低金属负载下,金属前驱体迅速附着在氧化物表面,经过适当处理后产生均匀分布的单原子结构。但此方法操作复杂,沉淀剂的加入时机、pH值和沉淀温度对所制备样品的形貌和性能影响较大,不易控制。
光化学/电化学沉积方法通过光或者电化学辅助还原载体表面上吸附的单核金属前驱体。电化学沉积法可以将负载量控制在比载体缺陷还低的水平。由于单个金属原子的电子状态不同,可以通过阴/阳极沉积以确定单个原子的价态,但该方法制备的催化剂产率低,在高电压下氧化物载体容易被重构或发生相变。光化学还原方法也是制备金属氧化物负载SACs催化剂的普适方法。例如, LIU等[22]通过光还原方法,利用光生电子在RuO2/CeO2莫特-肖特基结之间的传输和转移,选择性地将Pt还原在RuO2表面。光化学合成方法是一种温和条件下精准控制金属单原子生长位置的简单策略。
3 金属-氧化物SACs的表征
球差校正扫描透射电子显微镜(AC-STEM)是证明单个金属原子的重要工具,它能够直观地提供单原子物种局部结构信息,但不能反映样品的整体信息。而X射线吸收光谱(XAS)由于其高灵敏度特点可以提供单原子物种的宏观平均信息。此外,红外光谱(IR)也成为表征SACs的重要手段。电子顺磁共振光谱(EPR)可以提供金属氧化物上不饱和配位中心、SACs与氧化物载体之间的强相互作用信息。核磁共振波谱(NMR)可提供催化剂结构和吸附剂相互作用信息。
电子显微镜(EM)能更直观并以更少的样品消耗提供不同材料的各种信息,是表征固体催化剂结构和形貌的有力工具。由于EM技术的不断进步,亚埃分辨率的AC-STEM已成为高角度环形暗场(HAADF)成像模式下准确识别氧化物载体上单个金属原子存在和空间分布的基本工具。AC-STEM-HAADF可以直接观察到氧化物上/内异位单原子的位置,即氧空位和金属空位在氧化物载体表面上的具体位置,为深入研究SACs的结构-催化活性构效关系提供强有力的依据。此外,新开发的环形亮场STEM(ABF-STEM)与HAADF-STEM结合,有望检测出高原子序数氧化物(如La2O3、CeO2)上负载的轻元素金属原子。另外,(原位)环形透射电镜能够在气体或液体条件下进行实时监测,并逐渐成为原位检测原子动态的一种通用方法。WEI等[23]利用原位环形透射电镜记录了Pd、Pt和Au纳米粒子在900 ℃以上的惰性气氛中转变为热稳定单原子的动态过程。
XAS可用于研究原子和电子的结构,通常分为扩展X射线吸收精细结构谱(EXAFS)和X射线吸收近边结构光谱(XANES)。EXAFS光谱是通过确定吸收原子的配位数、键距离和相邻原子的种类来评估特定元素的原子环境,并提供金属-载体成键的证据。XANES对吸收原子的氧化态和配位状态非常敏感,可用于提取其电子信息。
一般来说,在转换至R空间后, EXAFS 光谱中若没有金属-金属壳层,则表明金属单原子在载体材料上孤立存在。对EXAFS光谱进行拟合分析,可以进一步获得被检测原子的配位状态和活性位点结构。XANES光谱能够证明负载单一金属原子的化学状态,一般而言,氧化物负载的SACs中的金属原子总是处于氧化态而不是金属态。XAS还可以动态监测单个原子的几何结构和化学状态的演变行为。PETERSON等[24]采用XAS表征技术,检测到CO氧化反应过程中Pd—O峰强度随着反应温度的升高而降低,同时Pd金属峰强度迅速增加。
傅里叶变换红外光谱(FTIR)是由分子转动、振动时能级跃迁所产生的吸收光谱,光谱范围一般涵盖400~4000 cm-1之间。反应过程的中间物质可以通过时间和温度分辨红外光谱来检测。表征多相催化剂表面最常用的红外探针是CO、 H2、 NO和C2H4, 其中CO是检测氧化物负载SACs的最常用探针。CO吸附的红外光谱(CO-IR)可以识别氧化物负载的单原子催化剂中孤立金属原子的种类和均匀性,特别是当金属团簇、纳米颗粒和单原子共存时,CO-IR可以方便地提供特异性位点信息。通过CO-IR识别氧化物担载的Pt、Rh、Pd、Ir等单原子已被广泛应用,而氧化物担载的Ni、Au、Cu、Ru、Ag等非贵金属单原子催化剂的红外检测及识别技术仍需探索。
电子顺磁共振(EPR)可以提供顺磁中心的性质、对称性、电子结构、价电子的变化以及顺磁中心与反应物的相互作用,其常见研究对象包括具有顺磁性质的过渡金属离子、自由基等。除此之外,EPR还可以检测金属氧化物上的不饱和配位中心。氧空位是金属氧化物中常见的不饱和配位中心,不仅可以锚定单个原子,而且能够提供特定的反应活性位点。因此,可以通过EPR检测单原子与氧化物载体之间的强相互作用。
核磁共振(NMR)可用于确定单个金属原子的锚定位置,有时作为XAS的补充,是获得催化剂结构及与吸附剂相互作用信息的有力工具。此外,反应物分子在单个金属原子上的吸附行为也可以通过NMR来跟踪。NMR技术在SACs的表征中发挥了重要作用,同时 SACs的发展也促进了NMR灵敏度的提高。
4 金属-载体间的相互作用
对于负载型金属催化剂,金属的尺寸和价态可以通过载体与金属之间的相互作用来调控。目前,稳定氧化物载体上单金属原子的几何结构特征可分为四种类型: (1)通过缺陷选择性锚定;(2)与表面官能团(如—O和—OH)配位;(3)孔道结构限域;(4)通过金属-载体强相互作用(Strong Metal-Support Interaction,SMSI)嵌入氧化物晶格中。
4.1 缺陷附着作用
氧化物载体上存在多种缺陷,如氧空位(Ov)、金属空位(Mv)、边缘、台阶、平台等。由于这些缺陷位点都能够捕获金属原子,因此确定金属原子的具体位置具有挑战性。
根据金属氧化物材料的化学性质,金属原子可以通过不同方式在其表面稳定存在。对于金属空位,原子分辨率成像和EXAFS表征是研究单个活性位点局部结构的有效工具。例如,在Fe2O3载体上沉积少量的Pt前驱体后,用AC-STEM技术对样品进行深入的研究。结果表明,Pt原子全部占据了Fe2O3表面的Fe位点,表明Pt被Fe空位所俘获[2]。当单个金属离子被金属空位捕获时,它们可以与周围的氧物种配位,形成催化活性物质。EPR被广泛应用于识别氧化物载体上的单个原子和氧空位之间的相互作用。金属物种可以通过增强电荷转移机制来稳定缺陷位置。然而,氧化物载体上低密度的缺陷位限制了高负载量金属SACs的制备。
4.2 氧化物表面官能团与SACs之间的相互作用
在氧化物载体的表面上,过量的氧离子可以通过金属—氧(M—O)或金属—羟基(M—OH)相互作用与金属原子配位。增加氧化物表面的羟基密度是一种降低负载金属原子迁移率的有效策略,主要通过三种方法实现。第一,将载体材料缩小到纳米级;第二,加入碱金属离子(如Na+、K+等)到氧化物载体上,通过形成氧配位的M1Ox物种来促进贵金属物种的原子级分散,该方法既适用于可还原的氧化物,也适用于不可还原的氧化物。此外,水热处理也是产生—OH的有效方法,原位形成的—OH与金属原子发生强相互作用,并使金属原子稳定在载体上,形成SACs。
4.3 多孔结构限域
将孤立的金属物种限制在氧化物材料的多孔结构内,如Fe2O3[25]、WOx[26]、TiO2[14]、沸石[27]等,通过抑制可能的聚集来保持金属物种高度分散在载体材料上。最近,多孔CaO-Al2O3材料被认为是SACs合适的载体材料[28]。特别是,12CaO·7Al2O3(C12A7)具有独特的交联笼状结构,由于匹配的尺寸和正电荷,孤立的金属阳离子被限制在C12A7空腔纳米结构中。在苛刻的还原条件下,氧化物的纳米多孔结构提供了一种限制效应,以提高SACs的金属-载体相互作用及其催化性能。
4.4 金属-载体强相互作用
SMSI是多相催化中的重要概念,该效应会导致界面电荷转移、金属结构改变、分子吸附调变等现象,最终对催化反应活性产生重要影响。然而,SMSI是否发生在SACs上尚不清楚。HAN等[29]通过光化学方法在TiO2纳米片上沉积Pt,并通过350 ℃煅烧去除保护剂制备出Pt/TiO2SACs。实验结果表明,Pt 纳米颗粒和Pt原子都表现出SMSI特征,如可逆抑制小分子的化学吸附、金属和氧化物载体之间的电荷转移。然而,单原子和金属纳米颗粒在SMSI方面的起源和所需条件不同。Pt原子的SMSI仅仅发生在较高还原温度下(600 ℃),而300 ℃ 氧化之后,Pt原子和Pt纳米颗粒上的CO吸附均恢复,表明SMSI对贵金属物种具有可逆吸附特性。该项研究为确定金属单原子或纳米颗粒是否是真正的反应活性位点提供了新思路。
4.5 SACs的活性中心
随着纳米材料科学和现代表征技术的快速发展, 研究者发现表面不饱和配位原子一般是催化反应的活性位点。对于纳米材料,一般通过控制纳米晶的尺寸、形貌、晶相、晶面等实现表面原子、电子结构和分布调控,从而提高其催化性能。当纳米晶的尺寸降低至单原子或原子团簇时,其能级结构和电子结构发生根本性改变。HAN等[30]报道了单原子Fe—N—C嵌入到三维N掺杂介孔碳高效电催化剂,在碱性电解质溶液中对氧还原反应(ORR)表现出较高的半波电位、电流密度、转换效率(TOF) 和质量活性。理论计算表明,N掺杂石墨提高了FeN4位点的本征活性、负载Fe和N产生高密度的活性位点、三维介孔碳之间的交联促进了电子和质子的传输,从而使得所制备的催化剂表现出较高ORR活性。HAN等[31]利用氟调控超薄碳纳米片上的单原子Ni—N4位点以实现高效电催化CO2还原。所制备单原子催化剂在较宽的电压范围内可以达到95%的法拉第效率,在-0.97 V(vs RHE)下CO2还原为CO的产率可达1146 mmol/(gcat·h)。原位衰减全反射红外光谱(ATR-IR)和理论计算结果表明,F掺杂调节了Ni—N4中心位置的电子构型,降低了CO2的活化能垒,有利于*COOH中间体的生成。由于SACs的独特结构和性质,其活性位点受配位环境、载体的表面原子结构和电子结构等影响,因而对其活性中心的认识仍存在诸多争议。
5 SACs在催化反应中的应用
与SACs催化相关的反应包括水煤气转换 (Water-Gas Shift Reaction,简称WGSR)、CO氧化、选择性氧化、选择性氢化、碳碳键偶联等。WGSR (CO+H2O→H2+CO2)是能源化工领域制取纯氢的重要方式之一。传统Cu-Zn催化剂存在许多缺点,如常温及低温活性低、高温耐久性差、经济和能源成本高等。近年来,人们对在不同载体上分散的Au和Pt SACs进行了深入研究,制备了Pt/TiO2[32]、 Pt/CeO2[33]、Au/TiO2[34]、Au/CeO2[35]和Au/La2O3[36]等SACs,并在WGSR反应中表现出优异的催化活性。除Pt型和Au型WGS催化剂外,研究者还制备了其他贵金属单原子催化剂,如Ir/FeOx[37]、Rh/TiO2[37]、Pd/FeOx[39]等。与传统块体催化剂相比,此类SACs表现出更加优异的催化反应活性。
CO选择性氧化(PROX)被认为是一种经济有效的氢气净化方法,可应用于新一代燃料电池汽车。为了满足质子交换膜燃料电池(PEMFC)富H2和无CO的合成气要求,所需催化剂必须能够高效且高选择性地氧化CO分子,从而尽可能避免H2的氧化反应。Pt和Au SACs是PROX反应常用的催化剂,Au SACs的催化活性和选择性较高,但其稳定性较差。而Pt SACs正好相反,其稳定性较好,但催化活性和选择性较低。选择性氧化反应经常被用于合成高附加值产品,如苯乙烯和乙炔[40]、醇[41]、苯胺[42]等。
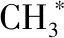
SACs的独特性质和优异活性, 使其在光催化领域有诸多运用。ZHOU等[45]利用阳极氧化法制备的TiO2纳米管阵列中的固有缺陷结构锚定了单原子Ir催化剂。与较高比表面积、特定晶面暴露的纳米TiO2相比,所制备的催化剂具有更加优异且稳定的光催化产氢性能。他们认为反应的活性中心是锚定在TiO2纳米管阵列表面缺陷中的Ir SACs, 其TOF值可以达到4×106h-1。SUI等[46]以SnO2为调节器,研发了一种锚定SACs(Pt、Cu和Ni等)的金属有机框架材料(MOFs),该方法不仅可以调控单原子金属位点,而且可以改变MOFs的结构,以UiO-66-NH2为例,Pt1/SnO2/UiO-66-NH2的光催化产氢速率是SnO2/UiO-66-NH2担载Pd纳米颗粒催化剂的5倍。另外,Pt1/SnO2/UiO-66-NH2的产氢速率可达2167 μmol/(g·h),远高于Cu1/SnO2/UiO-66-NH2和Ni1/SnO2/UiO-66-NH2的产氢速率。密度泛函理论计算(DFT)结果表明,单原子催化剂的产氢性能是由其与氢气结合能的差异所导致。
6 结语与展望
以载体和单原子之间的相互作用为切入点,简要介绍了氧化物负载的金属SACs,包括它们的分类、制备、表征以及在催化反应中应用。负载型SACs已经成为催化领域的研究前沿,相比于常规纳米催化剂,负载型SACs具有更高的催化活性、选择性和稳定性。另外,随着先进表征技术(同步辐射X射线吸收、球差电镜等)的发展,使得在亚埃级原子尺度上阐明SACs构效关系成为可能,同时也为多相催化与均相催化提供了机会。尽管如此,SACs结构的特殊性和复杂性使得该领域的研究仍面临很大挑战:
(1)活性原子的电子性质和相应的催化性能很大程度上由局部原子结构和配位环境决定,调节单原子活性中心的配位数和局部结构对氧化物负载的SACs尤其重要,但调变空间很小,因此精准调控配位原子及其数量的策略仍有待开发。
(2)虽然目前已报道了一些高负载和高稳定性SACs的合成方法,但关于大规模生产高效、稳定和高负载量SACs的报道仍然很少。因此,开发一种简单、普适性强的方法来实现SACs的大规模生产,对SACs的实际应用具有重要意义。
(3)对SACs相关的基本概念理解仍存在很大分歧,特别是真实反应条件下SACs的催化反应机理。因此,开发原位检测及更短时间分辨的表征仪器迫在眉睫,发展新型表征技术,对于单原子催化剂的合成和结构性能的优化具有重要意义。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
陶瓷学报(2020年6期)2021-01-26
当代陕西(2019年6期)2019-04-17
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
外语学刊(2014年3期)2014-12-03
郑州大学学报(理学版)(2013年2期)2013-03-11
